REPORTE DE CASO
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2019 - Universidad Ricardo PalmaDOI 10.25176/RFMH.v19i4.2145
SÍNDROME POEMS ASOCIADO A ENFERMEDAD DE CASTLEMAN
CASTLEMAN DISEASE VARIANT OF POEMS SYNDROME .
Pedro Paolo Sotelo-Jiménez1,a, Fanny Elizabeth Ramírez-Calderón1,b, María Del Pilar Quiñones-Avila1,c
1 Hospital Nacional Edgardo Rebagliati Martins, EsSalud, Lima-Perú.
a Médico Asistente Especialista en Medicina Interna.
b Médico Asistente Especialista en Emergencias.
c Médico Asistente Especialista en Anatomía Patológica.
RESUMEN
POEMS (Polineuropatía, Organomegalia, Endocrinopatía, Proteína Monoclonal y cambios en la piel) es un trastorno multisistémico raro. La enfermedad de Castleman es un trastorno linfoproliferativo atípico de causa desconocida, que puede estar asociado a POEMS.
Paciente mujer de 37 años de edad con cuadro clínico de 2 años de evolución caracterizado por múltiples adenopatías, adormecimiento, y debilidad de extremidades inferiores asociado a hiperpigmentación cutánea e hipertricosis. La electromiografía muestra una polineuropatía crónica activa, sensitiva-motora de tipo axonal; y la electroforesis sérica mostró banda monoclonal de Inmunoglobulina A tipo lambda. La biopsia de ganglio submandibular fue compatible con Enfermedad de Castleman.
Este reporte resalta el hecho que la enfermedad de Castleman se presenta en forma frecuente como parte de una variante del síndrome POEMS, y debe ser descartado en estos pacientes. Asimismo, la plasmaféresis puede ser útil en pacientes con síntomas neurológicos severos.
Palabras Clave: Sindrome de POEMS; Enfermedad de Castleman; Plasmaféresis (Fuente: DeCS)
ABSTRACT
POEMS (Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal Protein and skin changes) is a rare multisystemic disorder. Castleman's disease is an atypical lymphoproliferative disorder of unknown cause, which may be associated with POEMS.
A 37-year-old female patient with a clinical picture of 2 years of clinical evolution due to multiple adenopathies, numbness, and weakness of the lower extremities associated with cutaneous hyperpigmentation and hypertrichosis. Electromyography shows an active, sensory-motor chronic polyneuropathy of the axonal type; and serum electrophoresis showed monoclonal band of lambda type Immunoglobulin A. Submandibular ganglion biopsy was compatible with Castleman's disease.
This report highlights the fact that Castleman's disease occurs frequently as part of a variant of the POEMS syndrome, and should be ruled out in these patients. Also, plasmapheresis can be useful in patients with severe neurological symptoms.
Key words: POEMS syndrome; Castleman's disease; Plasmapheresis (Source: MeSH)
El síndrome POEMS (sinonimias: mieloma osteoesclerótico, síndrome Takatsuki, síndrome de Crow-Fukase) es un síndrome paraneoplásico raro asociado a discrasia de células plasmáticas, con una prevalencia reportada de aproximadamente 0,3 por 100,000 habitantes1,2, dándose con mayor frecuencia entre los 50 y 60 años. Una serie de 99 casos reportados en la Clínica Mayo3 reportó una edad promedio de 51 años (rango: 30-83), siendo el 63 % varones. El acrónimo POEMS fue establecido por Bardwick en 1980, el cual se refiere a las características del síndrome: poliradiculoneuropatía, organomegalia, endocrinopatía, desorden monoclonal de células plasmáticas y cambios cutáneos1-3. Hay tres puntos importantes relacionados con este acrónimo: 1) No se requieren todas las características para el diagnóstico; 2) Existen otras características no incluidas en el acrónimo, incluyendo papiledema, sobrecarga de volumen extravascular, lesiones óseas escleróticas, trombocitosis/eritrocitosis (PEST), así como niveles elevados del VEGF, predisposición hacia la trombosis y pruebas de función pulmonar alteradas; 3) Puede asociarse a un desorden clonal de células plasmáticas; y 4) Existe una variante asociada a la enfermedad de Castleman3 Dispenzieri sugiere un conjunto de criterios obligatorios, importantes, y menores2-4.
La enfermedad de Castleman (también conocida como hiperplasia ganglionar angiofolicular, hiperplasia linfoide gigante, hamartoma linfoide o linforreticuloma folicular) es un trastorno linfoproliferativo atípico de causa desconocida, caracterizado por crecimiento ganglionar hiperplásico no clonal4. En 1954 fue descrita por Benjamin Castleman, quien posteriormente la definió en una serie de 13 casos1. Se reconoce una mayor prevalencia en hombres con una relación 2:1, entre la quinta y sexta década de la vida3,4. En los Estados Unidos, se reporta una incidencia de 21 casos por millón de habitantes (3). Existen tres variedades histológicas: hialina vascular, de células plasmáticas, y mixta. Asimismo, existen dos formas clínicas: localizada y multicéntrica1,3. En la forma localizada hay solo un sitio de afección ganglionar: mediastino (60-75%), cuello (20%) o abdomen (10%); generalmente es asintomática o con síntomas relacionados al efecto de masa. Su tipo histológico más frecuente es la variedad hialina vascular y puede ser curable con la resección quirúrgica del tumor. La forma multicéntrica es menos común pero más agresiva. Se caracteriza por linfadenopatía generalizada y el tipo histológico más frecuente es de células plasmáticas. Generalmente se acompaña de síntomas sistémicos como fiebre, diaforesis nocturna, fatiga, anorexia, pérdida de peso. Esta forma requiere tratamiento sistémico y el rango de supervivencia es de 14-30 meses1,5.
La enfermedad de Castleman puede progresar a pancitopenia severa, falla multiorgánica, amiloidosis y linfoma; y puede estar relacionada al síndrome POEMS (6). Para ilustrar esta asociación presentamos el siguiente caso clínico.
CASO CLÍNICO
Paciente mujer de 37 años de edad, sin antecedente patológicos previos.
Presenta hace 2 años de evolución, sintomatología caracterizada por hiperpigmentación de pies-manos, pérdida de peso y amenorrea con una forma de inicio insidiosa y de curso progresivo por lo que acude a su policlínico de atención primaria de origen y se le diagnostica diabetes mellitus e hipotiroidismo, recibiendo tratamiento con 850mg de metformina y 100 ug de levotiroxina. Hace 4 meses se agrega adormecimiento, y debilidad de miembros inferiores (que impedía deambular) y superiores (caída de objetos de sus manos). Por estos síntomas acude a nuestra institución, donde es internada en el servicio de Neurología. Se realiza una electromiografía compatible con polineuropatía crónica activa, sensitivo-motora de tipo axonal, siendo transferida al servicio de Medicina Interna. Al examen físico luce adelgazada, se palpa adenomegalia submaxilar derecha y se evidencia hiperpigmentación de manos y pies, hipertricosis, leve acropaquia y cuadriparesia distal arrefléxica (Figura 1A). Se plantea la posibilidad de Síndrome POEMS.
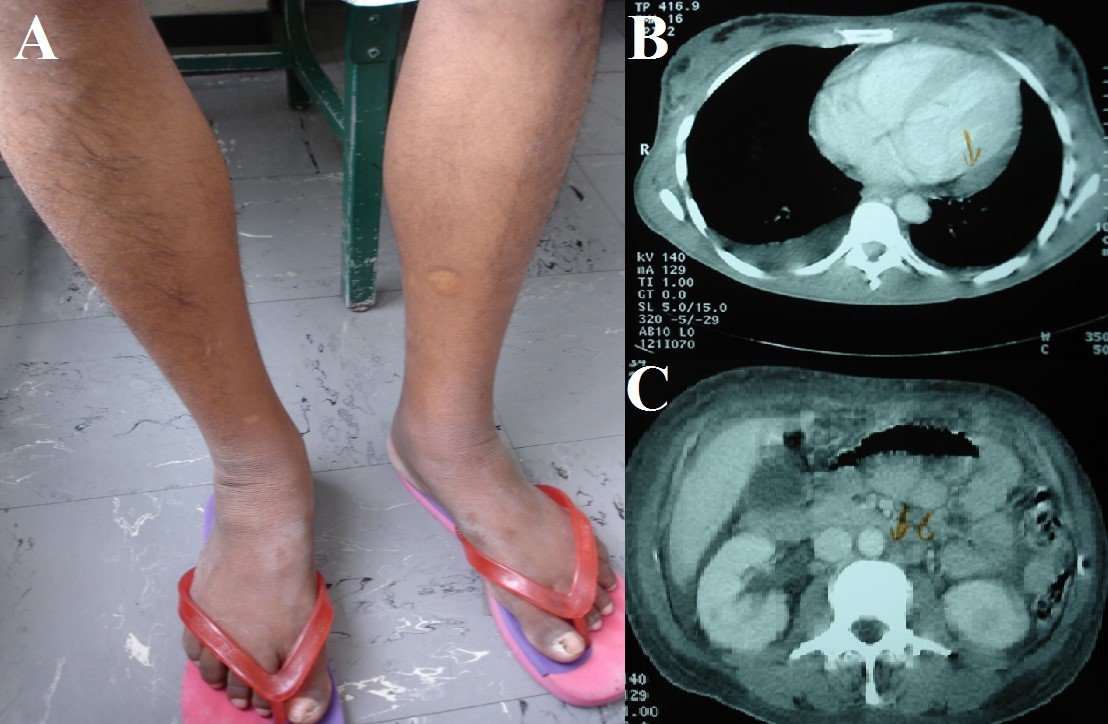
Figura 1. A: Hipertricosis e hiperpigmentación cutánea en piernas. B: Serositis (derrame pleural y pericárdico). C:Adenopatías intraabdominales.
La radiografía completa del esqueleto no mostró lesiones líticas ni blásticas. No hubo trombosis venosa profunda en la ecografía Doppler de extremidades inferiores. La TAC de torso mostró aspecto congestivo del tejido subcutáneo de pared torácica, abdominal y pélvica; liquido pericárdico 10 mm espesor; derrame pleural bilateral; leve hepatoesplenomegalia; riñones de aspecto congestivo; adenopatías retroperitoneales entre 10 y 15 mm (Figuras 1B y 1C). La electroforesis en sangre mostró leve incremento de gammaglobulinas; leve inversión en la relación Albúmina/Globulina, e Inmunoglobulina A 691 mg/dL (VN: 85-450). La electroforesis en orina con inmunofijación de cadenas ligeras evidenció pico monoclonal clase Ig A, tipo lambda (Figura 2A). Biopsia de piel mostró Hiperqueratosis ortoqueratósica, acantosis regular y papilomatosis. La médula ósea mostró hiperplasia megacariocítica e incremento de células plasmáticas en 10%. La Aspiración por Aguja Fina de ganglio submaxilar derecho mostró células aisladas con cambios atípicos. Finalmente, la biopsia quirúrgica de ganglio concluyó Enfermedad de Castleman variedad de células plasmáticas (Figura 2B). Pruebas para herpes virus 8 tanto en sangre como en tejidos no estuvieron disponibles.
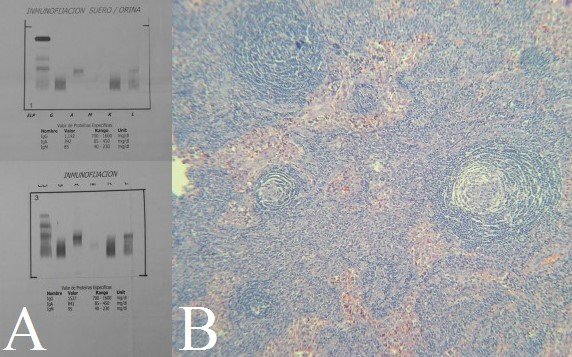
Figura 2. A: Banda monoclonal Ig A lambda en Inmunofijación de orina. B: Ganglio biopsiado mostrando folículo con patrón piel de cebolla.
DISCUSIÓN
Esta paciente ilustra un caso de POEMS cuya presentación inicial fue cuadriparesia, luego asociada a Enfermedad de Castleman. Solo hemos encontrado un reporte previo de esta asociación en nuestro país7 a pesar de estar estrechamente relacionados. Las series de casos más grandes reportan presencia de Enfermedad de Castleman multicéntrica en aproximadamente el 32-50% de los pacientes con POEMS2. Desafortunadamente, en este caso no se pudo establecer si la Enfermedad de Castleman estaba asociada a la presencia de Herpesvirus-8.
La neuropatía es el síntoma predominante del POEMS, y suele ser periférica, ascendente, simétrica; afectando la función motora y sensitiva, con presentación bilateral, progresión distal a proximal en las extremidades, observándose paresia severa en más de la mitad de los pacientes2,3,8 similar a la presentación de nuestra paciente. La organomegalia se manifiesta comúnmente como hepatomegalia, esplenomegalia y/o linfadenopatía2. La sobrecarga de volumen extravascular se manifiesta comúnmente como edema periférico; siendo comunes derrame pleural, ascitis y derrame pericárdico. Las manifestaciones cutáneas incluyen hiperpigmentación, pénfigo paraneoplásico, hipertricosis, rubor y acrocianosis. El papiledema está presente en al menos un tercio de los pacientes, probablemente en relación con el aumento marcado de las proteínas en LCR2,3.
La presencia de desorden monoclonal de células plasmáticas se requiere para el diagnóstico. La proteína M suele ser escasa y en muchos casos no supera los 3 gr/dL. La cadena pesada es usualmente IgG o IgA y las cadenas ligeras son de tipo Lambda. En el 15% de los casos no se detecta proteína monoclonal. Aproximadamente el 84% de los pacientes tienen endocrinopatía reconocida, con hipogonadismo como la anormalidad más común, seguido de anormalidades tiroideas, e insuficiencia adrenal. Las lesiones ósteo-escleróticas ocurren en aproximadamente 95% de los pacientes. La biopsia de médula ósea suele revelar hiperplasia megacariocítica. Un tercio de los pacientes no tienen células plasmáticas clonales en la biopsia; y el porcentaje de células plasmáticas observadas suele ser menor al 5%3. Los niveles de VEGF en plasma están marcadamente elevados y se correlacionan con la actividad de la enfermedad, además de asociación con herpesvirus tipo 82,3,5,9. Actualmente, un VEGF positivo puede usarse para realizar el diagnostico en pacientes con gammapatía monoclonal y neuropatía periférica desmielinizante2.
El algoritmo de tratamiento está basado en la extensión de infiltración por células plasmáticas. En los casos de pacientes con una lesión ósea aislada sin células plasmáticas clonales en médula ósea, dosis curativas de radiación es la terapia recomendada. En la forma diseminada, se recomienda tratamiento sistémico. Las recomendaciones de tratamientos se basan en series de casos e incluyen corticoesteroides ,lenalidomida, talidomida, rituximab,bortezomib, todos con efecto anti VEGF y anti TNF3,6. El trasplante autólogo de células madre produce una mejoría neurológica evidente dentro de los 6 meses, presumiblemente por regeneración axonal extensa y remielinización; esta terapia es considerada por algunos autores como tratamiento de primera línea para pacientes jóvenes con síndrome de POEMS, incluso con compromiso neurológico extenso6,9.
La quimioterapia con trasplante de células madre de sangre periférica también puede ser bastante eficaz. Las series de casos sugieren que el 100% de los pacientes logran al menos cierta mejoría neurológica, estas respuestas son duraderas, pero se han informado también recaídas. De los 59 pacientes con síndrome de POEMS tratados en la Clínica Mayo, la supervivencia fue de 98%, 94% y 75% a 1, 2 y 5 años, respectivamente10.
El pronóstico a largo plazo no es muy claro. Un estudio previo con 112 pacientes japoneses con síndrome de POEMS mostró una supervivencia media de 33 meses, siendo la mayoría de muertes debidas a falla multiorgánica, o asociadas a derrame pleural y ascitis intratable11. Una vez hecho el diagnóstico, los pacientes deben evaluarse exhaustivamente para definir una línea de base para futuras evaluaciones. El VEGF y las pruebas de proteínas monoclonales se deberían medir al finalizar la terapia, luego cada 3 meses, y posteriormente cada 6 meses. Las pruebas radiológicas y la evaluación neurológica se recomiendan inicialmente cada 6 meses seguidas de una vez al año. Para aquellos pacientes con respuesta hematológica completa y respuesta completa de VEGF y PET, las imágenes pueden realizarse con menos frecuencia2,4.
CONCLUSIÓN
La presencia de polineuropatía sensitivo-motora sin causa especifica en un paciente con lesiones dérmicas y serositis podría orientarnos a sospechar Síndrome POEMS. Este síndrome suele estar asociado a la Enfermedad de Castleman, por lo que siempre debería descartarse, ya que podría cambiar el manejo del paciente. La polineuropatía en nuestro paciente respondió adecuadamente a plasmaféresis, la cual debe ser considerada como alternativa de tratamiento en algunos casos, especialmente aquellos con compromiso neurológico severo.
Agradecimientos
Damos las gracias al Instituto de Evaluación de Tecnologías en Salud e Investigación (IETSI) de EsSalud por su apoyo en el desarrollo de este manuscrito mediante su programa de mentoría para publicaciones científicas y al Dr Luis Pereyra Caballero por su colaboración en el Diagnóstico y seguimiento del caso.
Contribuciones de autoría: Los autores participaron en la generación, redacción y aprobación final del caso clínico.
Financiamiento: Autofinanciado.
Conflictos de interés: Los autores declaran no tener conflictos de interés respecto al contenido de la publicación de este caso clínico.
Aspectos Éticos: Esta investigación cumple todos los principios éticos de la declaración de Helsinki. Se obtuvo el consentimiento de la paciente y de los profesionales involucrados en el caso.
Correspondencia: Pedro Paolo Sotelo Jiménez
Dirección: Domingo Cueto N°120, Jesús María. Lima, Perú
Teléfono: 2654901- anexo 3149 – 3415 Celular: 999675855
Correo electrónico: pepasoji@hotmail.com
REFERENCIAS BIBLIOGRÁFICAS
