REPORTE DE CASO
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2019 - Universidad Ricardo PalmaDOI 10.25176/RFMH.v19i4.2145
CASTLEMAN DISEASE VARIANT OF POEMS SYNDROME
SÍNDROME POEMS ASOCIADO A ENFERMEDAD DE CASTLEMAN
Pedro Paolo Sotelo-Jiménez1,a, Fanny Elizabeth Ramírez-Calderón1,b, María Del Pilar Quiñones-Avila1,c
1 Edgardo Rebagliati Martins National Hospital, EsSalud, Lima-Peru.
a Medical Assistant Specialist in Internal Medicine.
b Medical Assistant Emergency Specialist.
c Physician Assistant Specialist in Pathological Anatomy.
POEMS (Polyneuropathy, Organomegaly, Endocrinopathy, Monoclonal Protein and skin changes) is a rare multisystemic disorder. Castleman's disease is an atypical lymphoproliferative disorder of unknown cause, which may be associated with POEMS.
A 37-year-old female patient with a clinical picture of 2 years of clinical evolution due to multiple adenopathies, numbness, and weakness of the lower extremities associated with cutaneous hyperpigmentation and hypertrichosis. Electromyography shows an active, sensory-motor chronic polyneuropathy of the axonal type; and serum electrophoresis showed monoclonal band of lambda type Immunoglobulin A. Submandibular ganglion biopsy was compatible with Castleman's disease.
This report highlights the fact that Castleman's disease occurs frequently as part of a variant of the POEMS syndrome, and should be ruled out in these patients. Also, plasmapheresis can be useful in patients with severe neurological symptoms.
Palabras Clave: POEMS syndrome; Castleman's disease; Plasmapheresis (Source: MeSH).
RESUMEN
POEMS (Polineuropatía, Organomegalia, Endocrinopatía, Proteína Monoclonal y cambios en la piel) es un trastorno multisistémico raro. La enfermedad de Castleman es un trastorno linfoproliferativo atípico de causa desconocida, que puede estar asociado a POEMS.
Paciente mujer de 37 años de edad con cuadro clínico de 2 años de evolución caracterizado por múltiples adenopatías, adormecimiento, y debilidad de extremidades inferiores asociado a hiperpigmentación cutánea e hipertricosis. La electromiografía muestra una polineuropatía crónica activa, sensitiva-motora de tipo axonal; y la electroforesis sérica mostró banda monoclonal de Inmunoglobulina A tipo lambda. La biopsia de ganglio submandibular fue compatible con Enfermedad de Castleman.
Este reporte resalta el hecho que la enfermedad de Castleman se presenta en forma frecuente como parte de una variante del síndrome POEMS, y debe ser descartado en estos pacientes. Asimismo, la plasmaféresis puede ser útil en pacientes con síntomas neurológicos severos.
Palabras Clave: Sindrome de POEMS; Enfermedad de Castleman; Plasmaféresis (Fuente: DeCS)
POEMS syndrome (synonyms: osteosclerotic myeloma, Takatsuki syndrome, Crow-Fukase syndrome) is a rare paraneoplastic syndrome associated with plasma cell dyscrasia, with a reported prevalence of approximately 0,3 per 100,000 inhabitants1,2, with the highest frequency occurring between the ages of 50 and 60. A series of 99 cases reported in the Mayo Clinic3 reported an average age of 51 years (range: 30-83), being 63% males. The acronym POEMS was established by Bardwick in 1980, which refers to the characteristics of the syndrome: polyradiculoneuropathy, organomegaly, endocrinopathy, monoclonal plasma cell disorder and cutaneous changes1-3. There are three important points related to this acronym: 1) Not all features are required for diagnosis; 2) There are other features not included in the acronym, including papilloedema, extravascular volume overload, sclerotic bone lesions, thrombocytosis/erythrocytosis, as well as elevated levels of VEGF, predisposition to thrombosis and evidence of impaired lung function; 3) May be associated with clonal plasma cell disorder; and 4) There is a variant associated with Castleman3 Dispenzieri disease suggests a set of mandatory, important, and minor criteria2-4.
Castleman’s disease (also known as angiofollicular nodal hyperplasia, giant lymphoid hyperplasia, lymphoid hamartoma, or follicular lymphorreticuloma) is an atypical lymphoproliferative disorder of unknown cause, characterized by nonclonal hyperplastic nodal growth4. In 1954 it was described by Benjamin Castleman, who later defined it in a series of 13 cases1. A higher prevalence is recognized in men with a ratio of 2:1, between the fifth and sixth decade of lifetime 3,4. In the United States, an incidence of 21 cases per million inhabitants is reported (3). There are three histological varieties: vascular hyaline, of plasma cells, and mixed. There are also two clinical forms: localized and multicenter1,3. In the localized form there is only one place of nodal condition: mediastinum (60-75%), neck (20%) or abdomen (10%); it is usually asymptomatic or with symptoms related to the mass effect. Its most common histologic type is the hyaline vascular variety and can be curable with surgical tumor resection. The multicenter form is less common but more aggressive. It is characterized by generalized lymphadenopathy and the most common histologic type is plasma cells. It is usually accompanied by systemic symptoms such as fever, nocturnal diaphosis, fatigue, anorexia, weight loss. This form requires systemic treatment and the survival range is 14-30 months1,5.
Castleman’s disease may progress to severe pancytopenia, multiorgan failure, amyloidosis, and lymphoma; and may be related to POEMS syndrome (6). To illustrate this association we present the following clinical case.
CLINICAL CASE
A 37-year-old female patient with no previous pathological history presents 2 years of evolution, symptomatology characterized by hand-foot hyperpigmentation, weight loss and amenorrhea with an insidious and progressive onset form so she went to her primary care polyclinic of origin and was diagnosed with diabetes mellitus and hypothyroidism, receiving treatment with 850mg metformin and 100 ug levothyroxine. Four months ago numbness was added and weakness of lower (which prevented wandering) and higher (falling objects from his hands) limbs. For these symptoms she went to our institution, where she was admitted to the neurology service. Electromyography compatible with active, sensory-motor chronic polyneuropathy of the axonal type is performed and she was transferred to the Internal Medicine service. In the physical examination she looked slimmer, with right submaxillary adenomegaly and hand-foot hyperpigmentation, hypertricosis, mild acropaquia and distal arreflexic quadriparesia(Figure 1A). The possibility of POEMS syndrome is raised.
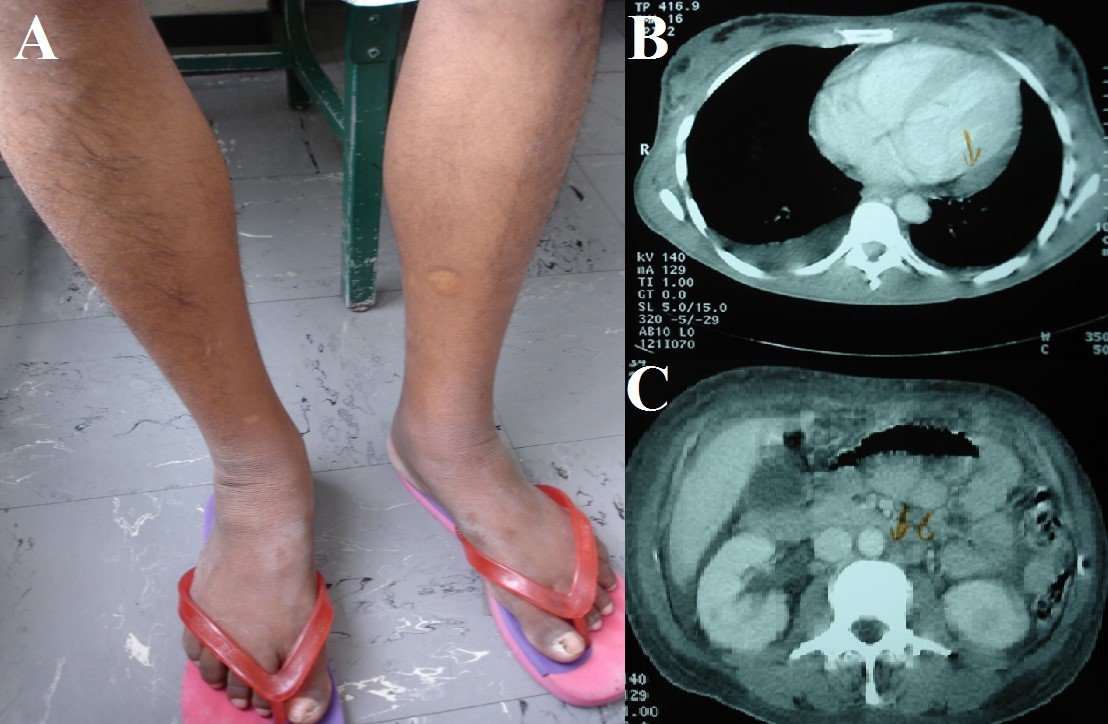
Figure 1. A: Hypertricosis and skin hyperpigmentation in the legs. B: Serositis (pleural and pericardial effusion). C:Intraabdominal adenopathies.
The complete X-ray of the skeleton showed no lytic or blastic lesions. There was no deep vein thrombosis in the Doppler ultrasound of the lower extremities. Torso CT showed congestive appearance of subcutaneous chest wall, abdominal and pelvic tissue; pericardial fluid 10 mm thick; bilateral pleural effusion; mild hepatosplenomegaly; congested-looking kidneys; retroperitoneal adenopathies between 10 and 15 mm (Figures 1B y 1C). Blood electrophoresis showed a slight increase in gamma globulins; a slight inversion in the ratio Albumin/Globulin, and Immunoglobulin A 691 mg/dL (VN: 85-450). Urine electrophoresis with light chain immunofixation showed monoclonal peak class Ig A, lambda type (Figure 2A). Skin biopsy showed orthokeratotic hyperkeratosis, regular acanthosis and papillomatosis. The bone marrow showed megakaryocytic hyperplasia and increased plasma cells by 10%. Right submaxillary ganglion fine needle aspiration showed isolated cells with atypical changes. Finally, surgical node biopsy concluded Castleman’s disease plasma cell variety (Figure 2B). Tests for herpes virus 8 in both blood and tissues were not available.
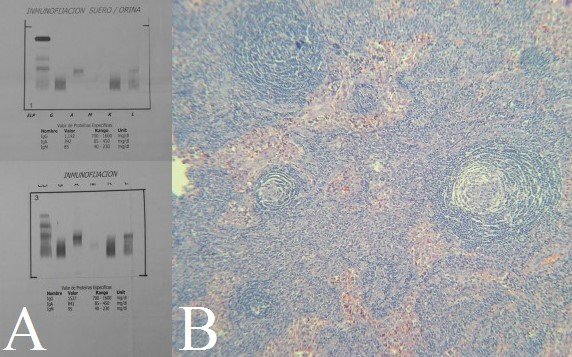
Figure 2. A: Ig A lambda monoclonal band in urine immunofixation.B: Biopsied ganglion showing follicle with onion skin pattern.
DISCUSSION
This patient illustrates a case of POEMS whose initial presentation was quadriparesis, later associated with Castleman’s disease. We have only found a previous report of this association in our country7despite being closely related. Larger case series report presence of multicenter Castleman disease in approximately 32-50% of POEMS2 patients. Unfortunately, in this case it could not be established whether Castleman’s disease was associated with the presence of Herpesvirus-8.
Neuropathy is the predominant symptom of POEMS, and is usually peripheral, ascending, symmetrical; affecting motor and sensory function, with bilateral presentation, distal to proximal progression in the extremities, severe paresis is observed in more than half of the patients2,3,8 similar to the presentation of our patient. Organomegaly is commonly manifested as hepatomegaly, splenomegaly and/or lymphadenopathy2. Extravascular volume overload is commonly manifested as peripheral oedema; pleural effusion, ascites and pericardial effusion are common. Skin manifestations include hyperpigmentation, paraneoplastic pemphigus, hypertricosis, flushing, and acrocyanosis. Papilloedema is present in at least one third of patients, probably in relation to the marked increase of proteins in LCR2,3.
The presence of monoclonal plasma cell disorder is required for diagnosis. Protein M is usually scarce and in many cases does not exceed 3 gr/dL. The heavy chain is usually IgG or IgA and mild chains are Lambda type. In 15% of cases monoclonal protein is not detected. Approximately 84% of patients have recognized endocrinopathy, with hypogonadism as the most common abnormality, followed by thyroid abnormalities, and adrenal insufficiency. Osmotic-sclerotic lesions occur in approximately 95% of patients. Bone marrow biopsy usually reveals megakaryocytic hyperplasia. One third of patients do not have clonal plasma cells in biopsy; and the percentage of plasma cells observed is usually less than 5%3. Plasma VEGF levels are markedly elevated and correlate with disease activity, in addition to association with herpesvirus type 82,3,5,9. Currently, a positive VEGF can be used to diagnose patients with monoclonal gammopathy and peripheral neuropathy demyelinate2.
The treatment algorithm is based on the extent of plasma cell infiltration. For patients with isolated bone lesion without clonal plasma cells in bone marrow, curative radiation doses are recommended. In the disseminated form, systemic treatment is recommended. Treatment recommendations are based on case series and include corticosteroids,lenalidomide, thalidomide, rituximab,bortezomib, all with anti VEGF and anti TNF effect3,6. Autologous stem cell transplantation produces an evident neurological improvement within 6 months, presumably by extensive axonal regeneration and remielinization; this therapy is considered by some authors as first-line treatment for young patients with POEMS syndrome, even with extensive neurological involvement6,9.
Chemotherapy with peripheral blood stem cell transplantation may also be quite effective. Case series suggest that 100% of patients achieve at least some neurological improvement, these responses are durable, but relapses have also been reported. Of the 59 patients with POEMS syndrome treated at the Mayo Clinic, survival was 98%, 94% and 75% at 1, 2 and 5 years, respectively10.
The long-term forecast is not very clear. A previous study of 112 Japanese patients with POEMS syndrome showed an average survival of 33 months, with the majority of deaths due to multiorgan failure, or associated with pleural effusion and intratable ascites11. After diagnosis, patients should be thoroughly evaluated to define a baseline for future evaluations. VEGF and monoclonal protein tests should be measured at the end of therapy, then every 3 months, and then every 6 months. Radiological tests and neurological evaluation are initially recommended every 6 months followed once a year. For those patients with complete haematological response and complete VEGF and PET response, imaging may be performed less frequently2,4.
CONCLUSIONS
The presence of sensory-motor polyneuropathy without a specific cause in a patient with dermal lesions and serositis could lead us to suspect POEMS syndrome. This syndrome is usually associated with Castleman’s disease, so it should always be discarded, as it could change the patient’s management. The polyneuropathy in our patient responded adequately to plasmapheresis, which should be considered as an alternative treatment in some cases, especially those with severe neurological involvement.
Special Thanks
We thank the Institute for the Evaluation of Technologies in Health and Research of EsSalud for his support in the development of this manuscript through his mentoring program for scientific publications and to Dr Luis Pereyra Caballero for his collaboration in the diagnosis and follow-up of the case.
Authorship contributions: The authors participated in the generation, writing and final approval of the clinical case.
Funding: Self-financed.
Conflicts of interest: The authors declare that they have no conflicts of interest with regard to the content of the publication of this clinical case.
Ethical Aspects: This research complies with all the ethical principles of the Helsinki Declaration. Consent was obtained from the patient and the professionals involved in the case.
Correspondence: Pedro Paolo Sotelo Jiménez
Address: Domingo Cueto N°120, Jesús María. Lima, Perú
Phone Number: 2654901- anexo 3149 – 3415 Cellphone: 999675855
Email: pepasoji@hotmail.com
BIBLIOGRAPHIC REFERENCES
