INTRODUCCIÓN
La amiloidosis, antiguamente llamada amiloidosis sistémica primaria, es una enfermedad producida por el depósito extracelular de proteínas insolubles con una estructura fibrilar compuesta de subunidades de bajo peso molecular, la mayoría de las cuales se encuentra entre 5 y 25 Kd, siendo condicionado por alteraciones en el plegamiento de estas proteínas. Tiene una tinción positiva para Rojo Congo y birrefringencia verde manzana en la microscopia de luz polarizada. Cursa con preponderancia en el sexo masculino. Los síntomas más frecuentes son fatiga y pérdida de peso; con respecto a los hallazgos físicos, en su mayoría no son característicos, siendo la macroglosia uno de los signos más específicos. Su presentación sindrómica compromete frecuentemente riñón, con proteinuria en rango nefrótico con o sin insuficiencia renal y corazón, con falla cardiaca congestiva por miocardiopatía restrictiva; también hay compromiso a nivel gastrointestinal y del sistema nervioso1,2,3,4.
Entre los tipos de amiloidosis, se describe el tipo primario y el secundario. En la amiloidosis primaria (AL), el depósito proteico deriva de fragmentos de cadenas ligeras de inmunoglobulinas (la porción variable), y se encuentra en el grupo de discrasias de células plasmáticas con expansión clonal. La forma secundaria (AA) ocurre como complicación de enfermedades crónicas como artritis reumatoide (AR), espondilo artropatías, osteomielitis y tuberculosis1.
REPORTE DEL CASO
Paciente varón de 47 años, admitido al servicio de nefrología del Hospital Almenara, con un tiempo de enfermedad de 1 año y 6 meses, caracterizada por orina espumosa, disminución de peso progresivo y edema de miembros inferiores. Es diagnosticado de enfermedad renal crónica (ERC) recibiendo tratamiento médico. Cuatro meses antes de su ingreso, inició terapia de hemodiálisis. Desde hace 2 meses cursa con rigidez de manos bilateral, que inicia en el primer dedo de ambas manos, extendiéndose hasta el tercero en forma progresiva con limitación para la actividad funcional, no reporta disestesias. Cursa con pérdida de peso ponderal 10 kilos aproximadamente en las últimas dos semanas. Al ingreso refiere disfagia sólo a líquidos, disartria, dolor en cavidad oral y a la deglución; persistencia de orina espumosa; niega edemas, vómitos, llenura precoz, diarrea y fiebre. No reporta antecedentes médicos de importancia, pero si antecedentes familiares, padre tenía diabetes y ERC, tía materna y primo materno con ERC estadio 5 en terapia de hemodiálisis (3 veces por semana) no precisaba la causa.
El examen físico al ingreso: presión arterial: 120/80 mmHg, frecuencia cardiaca: 80 lat/min., frecuencia respiratoria: 18/min., peo 53 kg, palidez, no edema, piel adelgazada con atrofia muscular. Cavidad oral con macroglosia. No compromiso pulmonar, cardiovascular ni presencia de visceromegalia. En el examen locomotor el tinnel fue negativo y en ambas manos había rigidez bilateral con limitación a la movilidad.
Los exámenes mostraron: Hemograma: leucocitos 9210/mm3, linfocitos 2210/mm3, abastonados 276/mm 3, monocitos: 276/mm3, Hb: 6.67 g/dl, VCM: 97 um3, HCM: 32 pg, viscosidad sérica 4´5”, creatinina: 7.16 mg/dl, urea: 56 mg/dl, glucosa 69 mg/dl, fosfatasa alcalina: 67 U/L, TGP: 13 U/L, albúmina: 3.42 gr/dl, VSG: 103 mm/H, plaquetas: 353 mil/mm3, fibrinógeno: 3.7 g/L, TTPA: 27” TP: 9.7” INR: 0.93. Examen completo de orina: Densidad 1000, pH 8, glucosa +++, thevenon +++, leucocitos 10-14 xc, hematíes 1-2 xc. Ác. Úrico: 6.68 mg/dl, fósforo: 1.97mmol/L, calcio total: 3.58 mmol/L, calcio iónico: 1.47 mmol/L, F.R.: 4 UI/ml, PCR: 12.5 mg/L, ferritina: más de 1500 ng/ml, Ca19-9: 2.93 U/ml, Ca15-3: 8.06, AFP: 3.37 ng/ml., CEA:0.99 ng/ml. B2 microglobulina: 31.6 mg/L, proteinuria en orina de 24 horas: 3186 mg, marcadores para hepatitis B y C negativos. Proteinograma electroforético en plasma con hipoproteinemia moderada e incremento de alfa1, alfa2 y beta y disminución de gamma. Proteinograma electroforético en orina con proteinuria marcada no selectiva con una inmunofijación positiva para Bence Jones.
Ecográficamente ambos riñones muestran hallazgos compatibles con ERC. En la TEM abdominopélvica no reportan tumores ni adenopatías. La endoscopía digestiva alta evidencia esofagitis eritematosa distal con signos indirectos de gastroparesia. La biopsia por aspiración de tejido graso subcutáneo con Rojo de Congo fue negativa. Durante su evolución el paciente cursó con una neumonía intrahospitalaria y fallece.
El diagnóstico patológico fue el de una amiloidosis sistémica con compromiso mediastinal, pericárdico, pulmonar y renal (Figura 1).
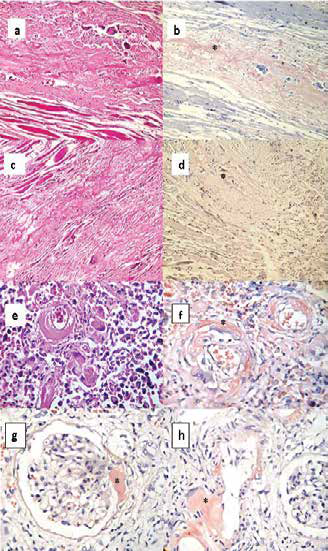 Figura 1.
Figura 1. Histopatología en hematoxilina-eosina y rojo Congo (+) (en asterisco) en: músculo pre laríngeo (a y b), pericardio (c y d), pulmón (e y f) y riñón (g y h). Se aprecia depósito de material hialino amorfo acelular y presencia de células gigantes multinucleadas tipo cuerpo extraño en fibras musculares (a), en pericardio, con áreas de fibrosis (c), en pulmones, a nivel pervivascular (e), y en riñones, en mesangio, polo vascular y vasos sanguíneos (g). Rojo Congo (+) (b, d, f, g y h).
DISCUSIÓN
El presente caso estuvo constituido por un paciente adulto, sin antecedentes relacionados a enfermedades crónicas, con antecedentes familiares de enfermedad renal crónica (línea materna), en la que se podría pensar alguna forma de amiloidosis heredofamiliar, cuyo desorden heterocigoto dominante, substituye un aminoácido proteico volviéndolo amiloidogénico, siendo la transtiretina la proteína amiloidogénica más común. El pronóstico de vida es más prolongado, a diferencia de este caso, que tuvo una sobrevida de 4 meses y había macroglosia, lo que generalmente no se describe en la forma familiar2,3,6.
La ausencia de enfermedades inflamatorias crónicas no apoyaba una forma de amiloidosis secundaria (AA). El paciente había iniciado hace 4 meses la terapia dialítica por lo que alejaba la posibilidad de una amiloidosis asociada a diálisis. Ésta se describe generalmente en pacientes con tratamiento dialítico mayor de 10 años, y es resultado del depósito de estructuras fibrilares derivadas de la beta 2 microglobulina, con presencia de signos típicos como el hombro doloroso por la infiltración amiloidea en la membrana sinovial en la articulación glenohumeral5 y, que no tenía el paciente. La forma clínica de este caso, más bien, se manifestó con enfermedad renal crónica evolutiva hasta el estadio 5, asociado a proteinuria en más de 3 gr, sin hipoalbuminemia severa, ni edema, y presentaba diuresis residual. En los pacientes con amiloidosis el riñón es el órgano más frecuentemente afectado. Un 75% cursa con proteinuria, y de ellos, el 30% en rango nefrótico. De los pacientes con amiloidosis renal, el 30% requerirá diálisis. Estimándose el tiempo promedio desde el diagnóstico hasta el inicio de diálisis de 14 meses, y el tiempo de sobrevida luego de iniciar la terapia dialítica de 8 meses1,2,6. En este caso, justamente existía el antecedente de manifestaciones clínicas de síndrome nefrótico 14 meses antes de iniciar la terapia dialítica, y la sobrevida luego de iniciar diálisis fue de 4 meses.
Se ha sugerido que existe un incremento de tamaño renal en la amiloidosis, como en efecto ocurre por deposito amiloideo, sin embargo, esta reportado que en la mayoría de pacientes el tamaño renal es normal 2,4, y en este caso se describían riñones hipotróficos asociados a la ERC. En relación a la discrasia de las células plasmáticas, en las gammapatías monoclonales como el mieloma múltiple, existe una relación usual de las cadenas ligeras kappa:lambda, la cual se invierte en la amiloidosis con una preponderancia de la cadena lambda en 3.5:1.
Sobre las manifestaciones extrarrenales de la amiloidosis, el segundo órgano mas frecuentemente afectado es el corazón, la cual se asocia a una miocardiopatia con disfunción diastólica. Cursa con un patrón de pseudoinfarto con complejos QS. La ecocardiografía es importante como ayuda diagnostica y puede describir un engrosamiento de la pared del miocardio ventricular sin antecedente de hipertensión, lo cual debiera hacer considerar un proceso infiltrativo como la amiloidosis. Este hallazgo no afecta la sobrevida, salvo se asocie a falla cardiaca o a un engrosamiento septal en más de 15 mm. Este paciente no se le realizó una ecocardiografía y no tuvo historia clínica de falla cardiaca, sin embargo, se debe considerar que la principal causa de muerte de los pacientes sometidos a terapia de sustitución renal son problemas cardiacos. A la microscopia se describió infiltración pericárdica, pero cabe recalcar que esto raramente resulta en pericarditis constrictiva 1,2,6.
El paciente no cursó con compromiso hepático (donde se describe una asociación con el aumento en la fosfatasa alcalina), no había hepatoesplenomegalia (se describe hepatomegalia en 25% de pacientes y la esplenomegalia es inusual). Había un síndrome consuntivo probablemente asociado a un síndrome de malabsorción. Cabe mencionar que en los pacientes con compromiso gastrointestinal, la mayoría cursan asintomáticos, describiéndose malabsorción asociada a diarrea en menos del 5%, por lo que no todos cursan con diarrea, asimismo, si bien se describe que generalmente existe la presencia de depósitos vasculares en la submucosa (lo cual no se describió en este paciente), los mecanismos asociados a malabsorción no están del todo establecidos ya que pueden incluir procesos como neuropatia autonómica del sistema mioentérico, miopatía secundaria a infiltración de músculo liso o isquemia secundaria a infiltración vascular1,2,6,7. Es importante remarcar que en este paciente se describió a nivel endoscópico signos indirectos de gastroparesia, probablemente asociado al compromiso neuropático.
Sobre el compromiso del sistema nervioso asociado a amiloidosis, el síndrome del tunel del carpo se presenta en cerca de la mitad de los pacientes, y con frecuencia se describe una neuropatía sensorial distal a proximal simétrica asociada en unas ocasiones a disestesias. Era sugerente en este paciente el compromiso del nervio mediano por la distribución sintomática del mismo. A nivel hematológico, tenía un perfil de coagulación normal, y no había trombocitosis (que se describe secundario a un hipoesplenismo en relación a una infiltración amiloidea). El compromiso a nivel de la coagulación generalmente se describe por deficiencia del factor X en relación a su ligazón con fibras amiloideas, disminución de factores de coagulación por compromiso hepático avanzado o por fibrinolisis alterada por deficiencia de la alfa 2 antiplasmina1,2,4,6,. El compromiso respiratorio en estos pacientes generalmente es asintomático, aunque en este caso existió una infiltración amiloidea, así como a nivel de la musculatura prelaringea.
Por último, en relación a la presentación clínica es importante diferenciar que en el caso de las amiloidosis AA, el componente cardiaco es inusual y pueden cursar con hepatoesplenomegalia. En el resto de enfermedades con depósito de inmunoglobulinas no amiloideas (enfermedad de depósito de cadenas pesadas, etc.), la progresión evolutiva es más lenta, y no ocurre macroglosia1,2,4.
El diagnóstico de este paciente se correlacionó con una proteinuria no selectiva, con imnunofijación para la proteína lambda de Bence Jones. En los pacientes con amiloidosis, el screening inicial de la proteína M (monoclonal), se debe realizar con la inmunoelectroforesis e inmunofijación en suero y orina, la cual será detectable en un 90% de casos. Se describe que la inmunofijación sérica tiene una sensibilidad de 69% versus la inmunofijación urinaria que tiene una sensibilidad del 83%, y que fue positiva en este paciente. En caso no se encuentre proteínas monoclonales por técnica de inmunofijación, se sugiere la cuantificación de cadenas ligeras libres, lo cual tiene una sensibilidad de 91% en los casos de amiloidosis AL. En pacientes con amiloidosis senil, familiar o localizada, no se documentan proteínas monoclonales4,6,8.
La biopsia por aspiración de tejido graso subcutáneo con rojo Congo tiene una sensibilidad de 57 a 85% y una especificidad de 92 al 100% tanto para la forma AA y AL. La biopsia rectal tiene una sensibilidad del 84%. La biopsia ósea es positiva en 30% de pacientes y la biopsia gingival en 50 a 70% de casos. La sensibilidad en general será mayor en compromiso multiorgánico4,6,8. La biopsia de tejido graso subcutáneo de este paciente fue negativa pese al compromiso multiorgánico, pero puede que la consunción marcada, probablemente no permitió una muestra suficiente de tejido graso. Finalmente, a nivel microscópico es importante saber que el amiloide AA y el AL pueden ser diferenciados por anticuerpos específicos. Tiempo atrás, se hacía un pretratamiento con permanganato de potasio, el cual elimina el rojo Congo positivo para el amiloide AA, más no para el amiloide AL 9.
Es así que para el diagnóstico de AL, se requerirán los siguientes 4 criterios: 1. Presencia de un síndrome sistémico relacionado al amiloide. 2. Tinción positiva rojo Congo en muestras como aspirado graso, médula ósea o biopsia de órgano. 3. Examen directo (tinción de inmunoperoxidasa, secuenciación) que revele al amiloide relacionado a cadenas ligeras. 4. Evidencia de expansión monoclonal celular (como proteína monoclonal sérica o urinaria, células plasmáticas en médula ósea)6.
Sobre el tratamiento se ha descrito el uso de inhibidores de proteosoma, drogas inmunomoduladoras y anticuerpos monoclonales y trasplante autólogo de células Stem, por lo que ha mejorado el pronóstico de los pacientes con un compromiso bajo a intermedio, sin embargo, para los de mayor compromiso el pronóstico aún es malo. El diagnóstico temprano será determinante para la efectividad del tratamiento y pronóstico, en razón a la progresión natural de la enfermedad3.
Contribuciones de autoría:El autor participó en la Concepción y diseño del trabajo; recolección / obtención de datos; contribución estadística; análisis e interpretación de datos; revisión crítica del manuscrito; redacción del manuscrito y aprobación de su versión final.
Financiamiento: Autofinanciamiento.
Conflictos de interés: El autor declara no tener conflicto de interés en la publicación de este artículo.
Recibido: 02 de diciembre 2018
Aprobado: 22 de enero 2019
Autor corresponsal: Edwin Rolando Castillo Velarde
Direccion: Av. Miguel Grau 800, La Victoria 15033. Lima-Perú.
Teléfono: +513242983
Correo electrónico: javiercdc@gmail.com
REFERENCIAS BIBLIOGRÁFICAS
2.- Gertz, Morie A. MD Amyloidosis. Monoclonal gammopathies and related disorders. Hematology, Oncology Clinics of North America. 1999; 13, Issue 6: 1211-33
3.- Vaxman I Gertz M, Recent Advances in the Diagnosis, Risk Stratification, and Management of Systemic Light-Chain Amyloidosis, Acta Haematol 2019; 141: 93–106.
4.- Gertz M, CME Information: Immunoglobulin Light Chain Amyloidosis: 2016 Update on Diagnosis, Prognosis, and Treatment, American Journal of Hematology 2016; 91 (9): 948-56.
5.- William L. Henrich. Diálisis. Amiloidosis por microglobulina B2 en la Nefropatía Terminal. 2º edición. México: McGraw Hill; 2001, p. 345-55.
6.- Leung, Nelson MD. Renal Manifestations of Plasma cell Disorders. American Journal of Kidney Diseases. 2007; 50 (1): 155-65.
7.- Suzanne R. Hayman, MD. Primary Systemic Amyloidosis: A cause of Malabsorption Syndrome. Am J Med. 2001; 111:535-40.
8.- Jacques Wallach. M.D. Interpretación Clínica de las pruebas de laboratorio. Amiloidosis. 4º edición. Barcelona: Masson; 2002. p. 1160-63.
9.- Sternberg's. Diagnostical Surgical Pathology. 4º edition, Phildadelphia: Lippincott Williams & Wilkins; 2004. p. 1905-09

http://bit.ly/2Jv5GAc
¿ Quieres dejar tu comentario o sugerencia sobre este articulo?
--> CLICK AQUI <---



