
3rd LATINOMARICAN CONFERENCE IN LIFESTYLE MEDICINE
INICIB

DOCENTES INVESTIGADORES EN REGINA

Latin American Lifestyle Medicine Association
CASO CLÍNICO
DOI 10.25176/RFMH.v19i3.2153
1 National Institute of Child Health San Borja, Lima-Peru.
2 Edgardo Rebagliati Martins National Hospital, Lima-Peru.
3 Research Institute in Biomedical Sciences (INICIB), Ricardo Palma University, Lima-Peru.
a Assistant doctor.
b Assistant physician pediatric cardiology unit.
c Occupational doctor.
d Researcher.
ABSTRACT
The present case corresponds to a 1-month-old male patient with a diagnosis of non-compacted cardiomyopathy associated with congenital heart defects. Noncompacted cardiomyopathy is newly included by the AHA as its own since the second half of the last decade. The diagnosis is mainly echocardiographic. Symptoms in children under one year may start with heart failure. The evolution is variable and tends to improve in some cases. Finally, in later decades, heart failure, thromboembolic events, malignant arrhythmias and sudden death become more pronounced. The management is in medicines for heart failure, to avoid malignant arrhythmias and thromboembolic events.
Keywords: Non-compacted cardiomyopathy, congenital heart defects, cardiac failure, echocardiography. (source: MeSH NLM)
RESUMEN
El presente caso corresponde a un paciente de 1 mes de vida, sexo masculino con diagnóstico de miocardiopatía no compactada asociada a defectos cardiacos congénitos. La miocardiopatía no compactada recién es incluida por la AHA como entidad propia a partir de la segunda mitad de la década pasada. El diagnóstico principalmente es ecocardiográfico. La sintomatología en menores de un año puede empezar con falla cardiaca. La evolución es variable con tendencia a la mejoría en algunos casos para finalmente en décadas posteriores se hace más pronunciada la falla cardiaca, eventos tromboembólicos, arritmias malignas y muerte súbita. El manejo esta en medicamentos para falla cardiaca, evitar arritmias malignas y eventos tromboembólicos.
Palabras clave:Miocardiopatía no compactada, defectos cardiacos congénitos, falla cardiaca, ecocardiografía. (fuente: DeCS BIREME)
INTRODUCTION
The non-compacted cardiomyopathy (NCM) represents a stop in the myocardial compaction process, characterized by the presence of multiple trabeculations and deep recesses between trabeculations, in addition to a thin compacted layer1.
This compaction process normally occurs between the fifth and eighth week of fetal life and is characterized by the gradual compaction of the myocardium, transformation of large intertrabecular spaces into capillaries and evolution of the coronary circulation. This process typically progresses from epicardium to endocardium and from base to apex2.
The true incidence of non-compacted cardiomyopathy is unknown as a result of diagnostic criteria and inconsistent nomenclature3. The prevalence of non-compacted cardiomyopathy is 0.01% in adults and 0.14% in children4.
Non-compacted cardiomyopathy has been identified as the most frequent after dilated and hypertrophic cardiomyopathy, representing 5-9% of cardiomyopathies in children 5 can be presented in isolation, associated with congenital heart disease, with genetic and hereditary component between 18 - 50%5.
Echocardiography is considered a reference for diagnosis. Chin et al in the 1990s gave the initial guidelines6 of a pathology poorly understood and included in the classification of myocardiopathies by the AHA from the second half of the last decade7.
The knowledge of this pathology is important to plan the management, prognosis and value the family study, besides assessing extracardiac pathology.
CASE REPORT
A previously healthy minor infant who, one day before admission, has poor lactation, irritability and respiratory distress, is admitted to a nearby hospital, finding a heart rate of 280, receives amiodarone and remits symptoms. The patient is transferred to the Hospital Edgardo Rebagliati Martins (HNERM) for study. Where he presents 2 episodes of supraventricular tachycardia (SVT), remitting with adenosine, being hospitalized in the Special Care Unit until hemodynamic stabilization. Clinical history: Birth weight 3308 g, gestational age 38 weeks, mixed lactation, weight gain of 29.7 g/d. Family history: Brothers with no history of heart disease. Healthy 38 year old father, 43 year old healthy mother. Physical examination: Weight: 4.2 kg, FC: 146, FR: 58, T: 36.6 ºC, PA: 85/65 mmHg, STO2: 98% (FIO2 21%). No dysmorphism, awake, ventilating spontaneously, capillary refill less than 3 seconds. Respiratory system: No rales. Heart system: Normodynamic precord, RCRR, SS II / VI EIB, palpable peripheral pulses of good intensity. Abdomen: Liver 2.5 cm below the right costal margin. Neurological: Fontanelle normotense, adequate tone and reflexes.
Supplementary tests
Figure 1Chest radiograph: ICT: 0.62, increased pulmonary flow.
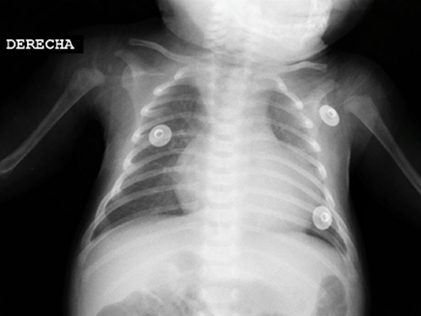
Figure 2. EKG: Sinus rhythm, HR: 150, PR: 120 ms, QRS: 60 ms, QTc: 440 ms, < QRS 0. Signs of left ventricular growth.

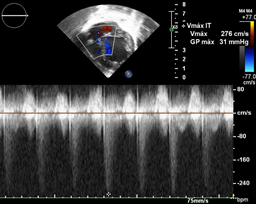
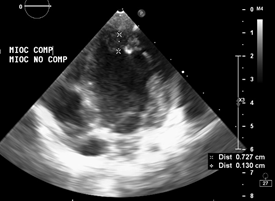
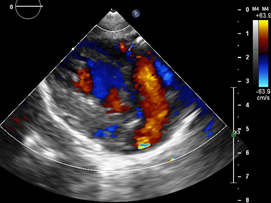
Echocardiography: DDVD: 16, DDSIV: 3, LVDD: 28, LVDD: 19 mm (+ 3DE), DPPVI: 3, R Ao: 11 mm, LA: 13 mm (0.05 DE), FE: 42% (Simpson), tricuspid regurgitation gradient 31mmHg (figure 5), radient V Ao 0.9 m / s (3 mmHg), PV gradient 1.3 m / s (8mmHg), pulmonary artery systolic pressure 36 mmHg mitral ring: 19.3 mm (+ 2DE), ring tricuspid: 20.7 mm (+ 2DE). Situs solitus, Levocardia, 4 veins drain in LA, suprahepatic collapses well, IVC (inferior vena cava) and SVC (superior vena cava) drain LA. AV agreement, tricuspid regurgitation and mild mitral regurgitation. VA concordance, competent sigmoid valves.
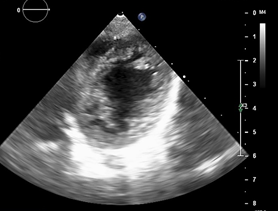
Left ventricle dilated, with SIV displaced to the right, more than 3 trabeculations at the level of the LV tip and the inferolateral and anterolateral wall (figures 6,7,8), with recesses between trabeculations and blood flow, myocardium not compacted / compacted from 5.5 (figures 9,10). Normal coronaries. 2 mm ventricular interventricular shunt from left to right (figure 3), 2 mm atrial septal defect shunt to left (figura 4), 1.5 mm PCA.
Glucose: 97 mg / dl, urea: 15 mg / dl, creatinine: 0.26 mg / dl, Hb: 12 mg / dl, platelets: 346 000, TP: 15, TTP: 49, troponin I: 0.373, Creatinin Kinase 192 U / l, CK -MB: 5.47 ng / ml, ESR: 19 mm / h fibrinogen: 238 mg / dl calcium: 10 mg / dl, phosphorus: 5.4 mg / dl, magnesium: 2 mEq / l, sodium: 138 mEq / l, K: 5 mEq / l, Cl: 109 mEq / l.



Definitive diagnosis:
Juvenile non-compacted cardiomyopathy, associated with congenital heart defects.
Differential diagnosis
Myocarditis.
Dilated cardiomyopathy.
Cardiomyopathy secondary to cardiac arrhythmia.
Medical management
Management of cardiac failure was initiated with diuretics, ACE inhibitor, beta-blocker and antiplatelet agent.
DISCUSSION
The non-compacted cardiomyopathy has been recognized as an entity separate from the other cardiomyopathies7. To date it is believed that it is due to a stop in the normal embryogenesis of the heart, with the arrest of the compaction process8. Knowing the etiology of the disease is found limited by genetic heterogeneity, with limited understanding of the regulation of myocardial trabeculation and compaction8.
To date it is not only considered that it would be caused by genetic alteration, family transmission has been found in 18-50% of adult patients, with phenotypic variability over time, being able to change to dilated or hypertrophic cardiomyopathy9. It has also been found the development of hypertrabeculation in young athletes10 and in pregnant women11 with subsequent reversion, considering that this disease could also be acquired.
Genetic studies have shown that the deficiency of the cytoplasmic protein FKBP 12 associated with the BMP / activin / TGFB1 receptors leads to poor regulation of the trabeculation and compaction of the ventricular myocardium8 and then leads to hypertrabeculation. To date, the genes involved in the non-compaction are FbKp1a, G4.5 / TAZ protein, deletion chromosome 14-3-3, protein ZASP, protein TNNT2, protein MYH7, protein TPM1, protein MYBPC3, protein ACTC1, among others12.
The case presented meets the echocardiographic criteria proposed by Jenni et all1, Chin el all6 y Stöllberger et all13 and more recently proposed by Lai et all3: Appearance of two layers of myocardium with thin compact layer and thick non-compact layer, increase in number of trabeculations lateral wall and apex VI, trabeculations that move synchronously with the myocardium, perfusion of intertrabecular recesses and abnormal ventricular function.
The age of presentation is variable, but with a greater number of diagnoses in children under one year of age and predominantly in males14 as in the case presented.
Clinical presentation ranges from asymptomatic15, heart failure, arrhythmias16, thromboembolic events and sudden death. Our patient presented with heart failure plus supraventricular tachycardia
The chest radiograph shows cardiomegaly and pulmonary hyperflow, not specific to this pathology. The EKG study shows signs of left ventricular growth, prolongation of the QT segment, data that are also reported in the literature16. The key diagnostic tool is echocardiography, however, it should be considered that no parameter is Gold Standard since they only value morphological parameters. The best views for the short axis and the apical 4 cameras are considered, but the other views should not be ignored. The most compromised segment is the ventricular apex, the other segments vary according to the study17; However, it must be considered that the more segments that are involved, the worse the prognosis18,19.
The functional assessment of the non-compacted myocardium in asymptomatic patients could be done by assessing the longitudinal strain by the speckle tracking technique20,21 and thus early determination of ventricular dysfunction and early treatment.
The management of patients with non-compacted cardiomyopathy isolated or associated with congenital heart defects, as in our patient, is aimed at managing extrapolated cardiac failure of dilated cardiomyopathy since there are no prospective studies in this regard12, avoiding systemic embolic events22 and prevention of malignant arrhythmias In the reported patient, management of heart failure and use of platelet antiaggregants was initiated.
The prognosis of these patients is variable, and it has been reported that ventricular dilatation, decreased ventricular function, age less than one year, dysmorphisms, among others constitute patients with worse prognosis23; although in another study, it has been mentioned that poor prognosis factors have an ejection fraction of less than 50 and hypoplasia of the posterior wall of the left ventricle24. According to the follow-up curves, pediatric patients who start with heart failure tend to improve over time, but in the end a progressive deterioration of cardiac function manifests itself, as does the adult, requiring even heart transplantation25.
Non-compacted cardiomyopathy is predominantly genetic, with variable clinical presentation, in some cases as the patient associated with congenital heart defects, which requires genetic counseling, genetic diagnosis and family cardiological screening26.
CONCLUSION
The NCM is increasing in recent years, it can be isolated or associated with congenital heart defects, the diagnosis is mainly by echocardiographic criteria; In the young child, it manifests with heart failure. Phenotypic expression can change to dilated or hypertrophic cardiomyopathy as phenotypic variability of the genes involved.
The family genetic load is generally autosomal dominant and predominates in the male sex. Management is extrapolated from dilated cardiomyopathy and is management of heart failure, prevention of malignant arrhythmias and systemic embolic events.
While at a younger age it is the diagnosis, less ventricular function and smaller compact wall of the left ventricle worse prognosis, requiring in some patients heart transplant.
