REPORTE DE CASO
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2019 - Universidad Ricardo PalmaDOI 10.25176/RFMH.v19i4.2345
CHILD MALIGINE OSTEOPETROSIS: A CASE REPORT AND LITERATURE REVIEW
OSTEOPETROSIS MALIGNA INFANTIL: A PROPOSITO DE UN CASO Y REVISION DE LA LITERATURA
Germán Aranzábal-Alegría1,
César Espinoza-Chiong1,
Dayanne Benites-Gamboa2,
Lino Aguirre Retamozo3
1 Research Institute in Biomedical Sciences, Universidad Ricardo Palma, Lima, Perú.
2 Faculty of Human Medicine, Universidad Ricardo Palma, Lima, Peru.
3 Hospital Nacional Dos de Mayo, Lima, Perú
ABSTRACT
Osteopetrosis comprises a series of rare genetic conditions that produce an imbalance in bone remodeling due to abnormal osteoclastic activity. We report a female patient 1 year 4 months, diagnosed with malignant osteopetrosis neuropathic child while causing paleness, distended abdomen with collateral circulation and growth retardation and developmental milestones are investigating. Examination right eye with retinopathy. Skeletal x-rays revealed a generalized bone hyperdensity. She bone marrow aspirate showed hypercellularity with hyperplastic erythroid, megakaryocytic normal white series. Diagnosis was confirmed by clinical and radiological features. The patient received supportive care. While the diagnosis is simple OPAR this is often delayed by the rarity of the disease and the lack of clinical suspicion, early diagnosis and treatment of bone marrow transplant is curative approach to a disease of poor prognosis.
Keywords: Osteopetrosis, infantile, osteoclast, retinopathy, bone (Source: Mesh).
RESUMEN
Osteopetrosis comprende una serie de raras condiciones genéticas que produce un desequilibrio en el remodelado óseo debido a una actividad osteoclástica anormal. Reportamos una paciente mujer de 1 año 4 meses, diagnosticada de osteopetrosis maligna infantil neuropática mientras se investigaba la causa de palidez, abdomen distendido con circulación colateral y retardo en el crecimiento y de los hitos del desarrollo. Examen oftalmológico con retinopatía derecha. Radiografías del esqueleto revelaron una hiperdensidad ósea generalizada. Aspirado de medula ósea mostró hipercelularidad con la serie eritroide hiperplásica, serie blanca y megacariocítica normal. Diagnóstico fue confirmado por características clínicas y radiológicas. La paciente recibió tratamiento de soporte. Aunque el diagnostico de osteopetrosis maligna infantil es sencillo este a menudo se retrasa por la infrecuencia de la enfermedad y la falta de sospecha clínica, un diagnóstico oportuno y un tratamiento de trasplante de medula ósea son el enfoque curativo para una enfermedad de pobre pronóstico.
Palabras Clave: Osteopetrosis, infantil, osteoclasto, retinopatía, hueso. (Fuente: Decs/Bireme).
Osteopetrosis comprises a series of rare genetic conditions characterized by an increase in bone density on radiographs1,caused by an abnormal osteoclastic activity that causes an imbalance in bone remodeling2,3.
The autosomal dominant form is usually asymptomatic frequently diagnosed in late childhood or adulthood and has a long survival time; However, the autosomal recessive form, also called infantile malignant osteopetrosis, is diagnosed in the first months of life at one year of age, with the worst prognosis4,5.The most severe manifestations tend to appear in childhood forms which include macrocephaly with frontal prominences, hydrocephalus, metabolic abnormalities, hematological abnormalities that include anemia, thrombocytopenia, extramedullary hematopoiesis and greater susceptibility to infections6,7. In addition, changes in the bone remodeling of the skull produce nerve compression generating blindness, deafness and facial paralysis8.
The case of a patient diagnosed with childhood malignant osteopetrosis presenting with early hematological and neurological complications is presented.
CLINICAL CASE
A 16 months-old female patient from Oxapampa who comes to the emergency department of Hospital nacional Dos de Mayo for fever, nasal congestion, rhinorrhea, cough and weight loss; He was given amoxicillin plus acetaminophen with the persistence of symptoms. she born of 35 weeks without complications with a weight of 2,900 gr, height of 47 cm and a cephalic perimeter of 34 cm; and one year old. She was hospitalized for 17 days for non-dysentery diarrhea, malnutrition and severe anemia of alleged nutritional versus parasitic origin; In addition, I have recurrent infections of the respiratory tract and gastrointestinal tract, so it was suggested transfer to Lima for further studies.
On physical examination, she presented general pallor, subcutaneous cellular tissue in a small amount, dystichiasis in the left eye, anisocoria, preserved photoreactivity, lateral subconjunctival hemorrhage in both eyes, anterior fontanel 4 cm, right nasal congestion, multifocal systolic murmur II / V, diffuse bilateral snoring, distended abdomen with collateral circulation and hepatomegaly (7 cm of the subcostal flange of normal consistency). Severe growth retardation and developmental milestones were observed (no cervico-thoracic control, language with monosyllables). In addition to presenting bilateral hearing loss.
Auxiliary test showed the following: Hb 4.5 gr / dl, leukocytes 22, 287 with a predominance of segmented (11, 145 / mm3), platelet count 20,000 / mm3, atypical lymphocytes 20%, blasts 10%. PCR 17.30 mg%, VSG 50 ml / h, TTP biochemical test 47.6 seconds, LDH 2053 IU / I, AFP 9.79 ng / m, Serum Calcium 8.7 mg / dl, TSH 1.32 µg, T4 0.7 µg, PTH 32.12 pg / ml, other tests within normal parameters. Urine test with red blood cells 12-15 / fields. Normal hemoglobin in electrophoresis. Direct Coombs test, serology for TORCH, HIV ELISA and Hepatitis was negative.
Chest x-ray, skull base and lower limbs showed a generalized bone hyperdensity (Figure 1,2,3,4). However, lumbar spine bone densitometry in normal ranges; Abdominal ultrasonography detected marked hepatomegaly and the cardiac test showed no structural alteration. Abdominopelvic multislice spiral tomography showed hepatomegaly with probable steatosis, no adenomegaly and constipation. Ophthalmoscopy showed optic neuropathy and retinopathy of the right eye. Medullary aspirate showed hypercellularity, M / E ratio: 1/1, severe hyperplasia erythroid series with orthochromatic predominance, normal myeloid series with maturation arrest with 4.5% blasts, normal megakaryocytic series, 8% lymphoid series and plasma cells 0 %. Normal cytogenetic study and evoked potentials were not performed due to poorly calibrated equipment at the time.
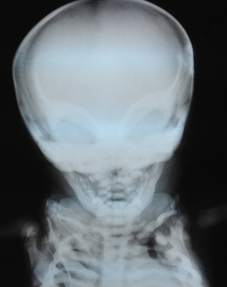
Figure 1. X-ray facial skeleton. Note the sclerosis of the sphenoid orbits and bone as "Harlequin's mask appearance."
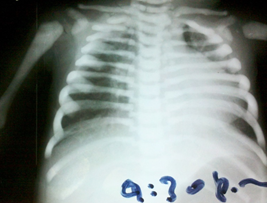
Figure 2. Chest x-ray. Notice the rickety rosary.
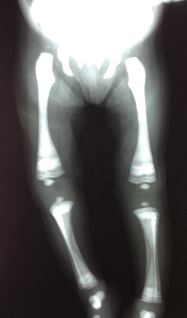
Figure 3. Radiography of the pelvis and lower limbs. Note the generalized bone density with the appearance of “bone inside the bone” or bone deformity in Erlenmeyer flask, easily recognizable in the femur and varus legs.
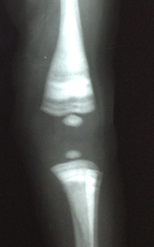
Figura 4. Distal femoral radiograph and left proximal tibial. Note the defects of metaphyseal modeling and the characteristic light bands.
Among the differential diagnoses raised were myelodysplastic syndromes, Paget's disease in bone presentation, hypoparathyroidism, pseudohypoparathyroidism and congenital infections. The definitive diagnosis was an infantile malignant osteopetrosis in the neuropathic variant due to the correlations of the radiographic and clinical characteristics; however, a genetic analysis could not be performed given the poor resources of parents. The patient received supportive and symptomatic treatment including antibiotic therapy for in-hospital pneumonia due to Staphylococcus epidermidis., Erythropoietin, blood transfusions and vitamin D and calcium supplementation. The patient went to home after 3 months of hospitalization with a big reference to the National Children's Institute for a bone marrow transplant, but after eight days she had a re-entry due to bronchial obstruction syndrome with favorable resolution, the case was not followed due upon transfer to another health center.
DISCUSSION
Osteopetrosis (OP), also called Albers-Schönberg disease or marble bone disease8, comprises a set of clinically different diseases that share the same radiological characteristic of an increase in bone density9-11.
This rare pathology lacks precise epidemiological data, but it is estimated that the incidence is less than 1-3.4: 200,000 births according to Del Fatorre et al 12. Its etiology is unknown although in animal models it was associated with an alteration in the formation and maturation of osteoclasts that become unable to perform bone and cartilaginous reabsorption generating an imbalance in bone remodeling13. The types of OP vary according to their severity, are divided into two large groups: 1. Autosomal recessive (OPAR) , 2.OP Autosomal dominant (OPAD) and 3. Linked to the X chromosome3.
The diagnosis of the OP is mainly made by the clinical-laboratory findings, the radiological findings being the ones that give us the diagnosis8, since there is no specific test for it12. The OPAR variety is classically presented during the first months of life14, as is the case with our patient, there is also a predisposition to suffer from complications such as nasal stiffness, related to paranasal sinus malformation, nerve compression neuropathies and pancytopenia8, evidenced in the patient's medical history. The clinical features of OPAR are the following: growth disorders, hydrocephalus-associated macrocephaly and frontal prominence, coanas stenosis, paranasal sinus disorders, hematological abnormalities compatible with spinal cord aplasia, neurological alterations such as blindness, deafness and facial paralysis associated with a stenosis of the foramen where these nerves and dental defects characterized by the presence of severe caries14. Of the clinical features mentioned, our patient presented an alteration in growth and development milestones, in addition to neurological alteration such as hearing loss bilateral, optic neuropathy and right retinopathy; Auxiliary test showed anemia, thrombocytopenia, leukocytosis with left deviation, 10% blasts and hepatomegaly discarding a myelodysplastic origin. Studies report masking and association of childhood malignant osteopetrosis and leukemias; however, no compatible findings were found in the bone marrow aspirate of our patient15.
The typical radiographic characteristics of osteopetrosis include a marked increase in bone density, which can be observed in figure 1,2,3,4 but in the initial radiology reports of our patient they were classified as normal, causing a delay in the diagnosis. In the OPAR there are subtypes such as: 1. Neuropathic OPAR, 2.OPAR plus renal tubular acidosis15, by the clinic presented by our patient is in the neuropathic variety characterized by neuropathy and hearing loss despite not having a neurophysiological evaluation of the hearing. It is important to make this difference because each type has a different response to the treatment and degree of relapse8.
Currently there is no effective treatment against this pathology8, the management offered to the patient is to provide support according to the symptomatology that is presenting11 during the natural course of the disease, our patient was offered a treatment for Inhospitable pneumonia infection that was the reason for prolonged hospitalization and treated with antibiotics for 14 days showing improvement, in addition to a decrease in the red and megakaryocytic series, hematinic began to be administered with which he was discharged.
Having a knowledge of the pathophysiology, the etiology of this disease cannot be specified in addition to a treatment to avoid the severity of symptoms such as optic nerve compression and vestibular-cochlear neuropathy9. The OP has a prognosis associated with the severity of the picture, in the infantile forms the children die as a result of the suppression of the bone marrow6 and recurrent infections. Currently, hematopoietic progenitor transplantation represents a therapeutic alternative whose response will depend on the type of underlying genetic mutation5. Although the diagnosis of childhood malignant osteopetrosis is simple and depends on a radiological and clinical-laboratory methods. The rarity of the disease and the lack of clinical suspicion, according to Essabar et al4, genetic studies have relevance to give counseling about genetic test to the parents of patients suffering from osteopetrosis, but were not carried out for our patient, given its high cost. Early diagnosis and timely supportive treatment prolongs the life of these patients that would otherwise be fatal. It is suggested develop more studies in animals in order to discover new therapeutic targets that improve the quality of life of patients.
THANKS
Francisca Valero-Villaízan, for her support in the literature search.
Author contributions: Espinoza-Chiong C, Aranzábal-Alegría G y Benites-Gamboa D; contributed in writing and final review of the clinical case, Aguirre-Retamozo L. contributed to the final review of the clinical case.
Funding Self-financed
Conflicts of interest: LThe authors declare no conflict of interest.
Correspondence: German Aranzábal-Alegría
Adress: Avenida Colombia 247, Lima 21, Perú.
Phone: 984767905
Email: garanzabal456@gmail.com
BIBLIOGRAPHY
