ARTICULO ORIGINAL
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2020 - Universidad Ricardo PalmaDOI 10.25176/RFMH.v20i4.2946
BASES GENÉTICAS DE LA HIPERTENSION ARTERIAL PULMONAR
GENETIC BASES OF PULMONARY ARTERIAL HYPERTENSION
María del Carmen Castro-Mujica1,a, Hugo Abarca-Barriga1,a
1Facultad de Medicina Humana – Universidad Ricardo Palma, Lima-Perú
aMédico Genetista
La Hipertensión arterial pulmonar (HAP) es una enfermedad heterogénea donde los genes juegan un rol sumamente importante. La HAP hereditaria (HAPh) se define como una condición genética de patrón de herencia autosómico dominante, de penetrancia incompleta, de expresividad variable, que presenta un fenómeno de anticipación y que agrupa a los casos de HAP familiar definido por la presencia de dos o más miembros de la familia con HAP con o sin variante germinal identificada y a los casos de HAP idiopática que corresponde a los casos aislados en la familia con una variante germinal identificada. Para establecer el diagnóstico de HAPh, es necesario confirmar el diagnóstico en al menos dos familiares (HAPf) o identificar la variante germinal en un caso aislado en la familia (HAPi).
Palabras Clave: Hipertensión pulmonar, asesoramiento genético (fuente: DeCS BIREME).
ABSTRACT
Pulmonary arterial hypertension (PAH) is a heterogeneous disease where genes play an extremely important role. Hereditary PAH (hAPH) is defined as a genetic condition with an autosomal dominant inheritance pattern, incomplete penetrance, variable expressiveness, presenting an anticipatory phenomenon and grouping cases of familial PAH defined by the presence of two or more members of the family with PAH with or without identified germline variant and idiopathic PAH cases corresponding to isolated cases in the family with an identified germline variant. To establish the diagnosis of hAPH, it is necessary to confirm the diagnosis in at least two relatives (fHAP) or to identify the germline variant in an isolated case in the family (HAPi).
Key words: Pulmonary hypertension; Genetic counseling (Source: MeSH NLM).
Inicialmente, la hipertensión arterial pulmonar (HAP) fue catalogada como HAP primaria, durante los años 1950 a 1998, donde el diagnóstico se realizaba por exclusión tras determinar la ausencia de causas identificables o factores de riesgos conocidos(1).
La HAP hereditaria (HAPh) es un término recientemente adoptado y que se define como una condición genética compleja, con patrón de herencia autosómico dominante (AD), con penetrancia incompleta influenciada por el género, de expresividad variable, y que presenta un fenómeno de anticipación.
La HAPh incluye a los casos de HAP familiar (HAPf) definido por la presencia de dos o más miembros de la familia con HAP con o sin una variante germinal identificada y a los casos de HAP idiopática (HAPi) que corresponde a los casos aislados en la familia con una variante germinal identificada(1-3). Estos últimos casos, podrían corresponder a casos en los que la enfermedad aun no se ha manifestado debido a la baja penetrancia o a variantes de novo. La HAPi corresponde aproximadamente al 94% del total de casos de HAP, mientras que el 6% restante corresponde a los casos de HAPf(4), siendo ambas clínica e histopatológicamente indistinguibles(1).
El gen BMPR2 (del inglés, bone morphogenetic protein receptor 2), que codifica el receptor 2 de la proteína morfogenética del hueso (BMPR-II) y que pertenece a la superfamilia del factor transformante del crecimiento beta (TGF-B), ha sido identificado en el 70-75% de los casos de HAPf y en el 10-40% de los casos de HAPi, siendo considerado el principal determinante genético de HAP(1). Existen otros genes involucrados en la patogénesis de HAP como ACVRL1, CAV1, KCNK3, ENDG, SMAD9, BMPR1B, entre otros, pero que son menos frecuentes (1-3%)(5).
La HAPh posee una mayor severidad de la enfermedad en los pacientes portadores de variates en el gen BMPR2 comparado con los no portadores de mutación, los cuáles se asocian a variantes “missense” en el gen BMPR2(6-8). Además se han identificado casos de HAP portadores de variantes en el gen BMPR2 que presentan fenómeno de anticipación, definido como la presentación de la enfermedad a una edad más temprana en las siguientes generaciones(3,9), sin embargo aún no existen estudios sistemáticos de base demográfica que se hayan realizado para evitar el sesgo de evaluación en estos casos(10).
Actualmente, el conocimiento de la genética y de la biología molecular nos permite entender los mecanismos y vías moleculares involucradas en la HAPh, siendo el objetivo de esta revisión brindar los alcances necesarios para su mejor comprensión para poder brindar al paciente un diagnóstico genético adecuado, un tratamiento especializado y sobretodo la asesoría genética correspondiente lo que permite informarle sobre los estudios genéticos-moleculares disponibles para la identificación de los portadores de mutación germinal y poder realizar un manejo y seguimiento individualizado del paciente y sus familiares portadores asintomáticos en riesgo.
HAPf Y EL DESCUBRIMIENTO DEL GEN BMPR2
La HAPf fue descrita por primera vez en 1951 por Dresdale et al(11). En el año 1997 mediante un estudio de ligamiento, usando marcadores microsatélites distribuidos a lo largo del genoma, que requería de un gran número de familias de pacientes y de controles para la comparación de las frecuencia de los genotipos en ambos grupos y determinar la asociación entre la enfermedad y el marcador, se identificó el locus 2q31-32 como región donde se localizaría al gen causante de HAPf(12,13). Posteriormente, en el año 2000, los genes candidatos fueron secuenciados y el gen BMPR2 fue identificado como la principal causa genética de HAPf(14-16). La búsqueda de genes candidatos relacionados a la via de señalización del gen BMPR2 continua siendo motivo de estudio para el entendimiento de la patogénesis molecular de HAPf(17).
Tras su descubrimiento en los casos de HAPf, el gen BMPR2 se convirtió en el gen candidato para los casos de HAPi ya que los fenotipos eran idénticos, lo que llevó a realizarse un estudio de 50 pacientes con HAPi, donde 13 de ellos (25%) eran portadores de variantes en el gen BMPR2(18). La HAPi, aparentemente esporádica, correspondiente a los casos aislados en la familia, sin antecedentes familiares, albergan variantes germinales en el gen BMPR2 en el 10-40% de los casos, por lo que constituyen un riesgo de transmisión hereditaria a desarrollar HAP en otros miembros de la familia; mientras que la HAPf, con antecedentes familiares con o sin variantes germinales identificadas, presentan variantes germinales en el gen BMPR2 en el 70% de los casos(16,19-22).
Actualmente con el desarrollo de nuevas tecnologías, como el secuenciamiento de próxima generación, podemos identificar a gran escala, las variantes comunes en diversos genes que estarían involucrado en la predisposición genética a desarrollar HAP.
CLASIFICACION DE HAP
La clasificación de la Hipertensión Pulmonar (HP) ha ido variando con el pasar de los años. Asi tenemos que la primera clasificación fue propuesta en 1973, donde se designaron las categorías de hipertensión pulmonar primaria (HPP) e hipertensión pulmonar secundaria (HPS), según la presencia o ausencia de factores de riesgo identificados(23,24). En el año 1998, durante el Segundo Congreso Mundial de Hipertensión Pulmonar (HP) realizado en Evian, Francia, se propuso una clasificación de la HAP basada en los datos clínicos de los pacientes a fin de individualizar las categorías que compartían similitudes en la patogénesis, características clínicas y opciones terapéuticas(25).
Posteriormente, en el año 2003, durante el Tercer Congreso Mundial de HP en Venecia, Italia se propusieron cambios importantes como el de eliminar el término “Hipertensión Pulmonar Primaria” e incorporar los términos “HAP idiopática” y “HAP familiar”(26) y en el año 2008, durante el Cuarto Congreso Mundial de HP en Dana Point, EEUU, el grupo internacional de expertos realizó la revisión de la clasificación previa y se propuso que se reemplazara el término “HAP familiar” por “HAP hereditaria”(2) y que el grupo 1 incluyera cinco subgrupos correspondientes a la HAPi, HAPh, HAP inducido por drogas y toxinas, HAP asociado a otras enfermedades y a la Hipertensión pulmonar persistente del RN(27,28). En la última clasificación de HAP, establecida en el año 2013 durante el Quinto Congreso Mundial de HP en Nice, Francia se propusieron algunas modificaciones mínimas sin alterar la estructura establecida previamente(19).
HAP EN NIÑOS
La HAP en niños es más severa y refractaria al tratamiento, y posee diferencias en su presentación ya que se han descrito dos picos de edad al diagnóstico, uno antes del primer año de vida y el próximo a los doce años de edad aproximadamente(29,30). Los pacientes que portan variantes en el gen BMPR2, presentan la alteración genética desde su concepción, sin embargo aún no se explica porque algunos casos la enfermedad se desarrolla en niños y en otros casos en adultos, lo que hace suponer que existirían otros factores que influirían en la edad de presentación de HAP(31,32).
La HAP en pacientes pediátricos es más heterogénea que en adultos y puede estar asociado a diversos síndromes genéticos, especialmente aquellos síndromes con enfermedad congénita cardiaca, enfermedad vascular y enfermedad hepática(10).
La HAP en niños tiene múltiples etiologías genéticas en comparación a la HAP en adultos y aunque es una complicación poco frecuente de algunos síndromes genéticos, puede encontrarse en síndrome Down y en otros síndromes genéticos pero que no necesariamente se asocien a enfermedad cardiaca congénita e HAP, como en síndrome DiGeorge, VACTERL, CHARGE, Noonan entre otras(10,17). Otros síndromes genéticos asociados a HAP, pero que usualmente no se encuentran asociados a enfermedad cardiaca congénita incluyen al síndrome de Adams-Oliver, neurofibromatosis 1, síndrome QT largo, sardiomiopatía hipertrófica, síndrome Cantu, snfermedad Gaucher y enfermedades de depósito de glucógeno(10,17).
La HAPh posee un patrón de herencia autosómico dominante, con penetrancia incompleta donde aproximadamente sólo el 20% de los portadores de una variante germinal en el gen BMPR2 van a desarrollar la enfermedad, lo que sugiere que la variante génica es necesaria pero no suficiente para expresar la enfermedad y que tal vez requiera otros factores génicos y/o ambientales que contribuyan a su desarrollo(5,29,33).
El sexo femenino es el factor mejor demostrado que influye en la penetrancia de HAP, con una relación mujer:varon de 3:1 en HAP de distintas etiologías, incluyendo HAPi, HAPf y HAP asociada a otras patologías (HAPa)(8,14,34), además se estima que la penetrancia varía según el género, siendo aproximadamente 10% en varones y 30% en mujeres(35).
La predominancia en mujeres, la penetrancia reducida y la edad al diagnóstico variable de HAP, ha sido motivo de estudio y diversas publicaciones han sugerido que el metabolismo del estrógeno es pieza clave en la penetrancia de HAP(35-37) por lo que se han comparado los aspectos clínicos entre los pacientes con HAP portadores de variantes en el gen BMPR2 y pacientes sin mutación identificada, según el género.
La mayoría de mujeres metaboliza estradiol (E2) en 2-estrogeno (2-OHE) y algunas metabolizan el E2 a 16-estrogeno (16a-OHE) lo que estimula la proliferación celular por activación constitutiva de los receptores de estrógenos(35). Además de ser más mitogénico que 2-OHE, la 16a-OHE puede ser más genotóxica y las pacientes que metabolizan una gran proporción de E2 en 16a-OHE incrementan su riesgo a desarrollar la enfermedad debido a estos efectos(36). En las pacientes portadoras de variantes en el gen BMPR2, el desarrollo de la enfermedad se relaciona fuertemente con la disminución de la relación 2-OHE/16a-OHE, asociado a una disminución en la expresión del citocromo que metaboliza los estrógenos, Citocromo p450 1B1 (CYP1B1), y a polimorfismos en éste(36). Algunos estudios han demostrado una disminución significativa de los niveles de los transcriptos del CYP1B1 en mujeres afectadas portadoras de variantes en el gen BMPR2 comparado con las no afectadas portadoras(37). Se ha propuesto que los estrógenos son posibles modificadores de la enfermedad debido a la menor prevalencia en mujeres pre púberes(38), además de ser potentes mitógenos de las células músculo liso de la vasculatura pulmonar(39). Otros estudios también han propuesto que los polimorfismos del gen TGFb1 causan desequilibrio en la vía de señalización TGF-B, influenciando en la penetrancia de las mutaciones BMPR2 y modulando la edad al diagnóstico de HAP(1,36,40).
FENÓMENO DE ANTICIPACIÓN EN HAP
El fenómeno de anticipación genética, es decir, el desarrollo de HAPf a una edad más temprana y de mayor severidad en las siguientes generaciones(41,42), ha sido evidenciado en diversos estudios; sin embargo no han podido ser explicados por fenómenos de expansión de trinucleotidos, ni acortamiento progresivo del teloméro como sucede en otras enfermedades(43), por lo que tal vez sean necesarios estudios de seguimiento a largo plazo para realizar las comparaciones apropiadas y con significancia estadística(44)
DIAGNOSTICO GENÉTICO-MOLECULAR EN HAP
La aplicación de la biología molecular en el estudio de los casos de HAPf, nos permite establecer su diagnóstico genético-molecular, así como aportar nuevos conocimientos en la investigación sobre su patogénesis.
El gen BMPR2, codifica a la proteína BMPR-II que se expresa en las células del endotelio pulmonar y células del músculo liso principalmente, regulando múltiples funciones como la proliferación, migración, diferenciación y apoptosis celular, y es además un miembro de la superfamilia de receptores del TGF-ß(41). Los miembros de la superfamilia TGF-ß se subdividen en varias familias, que incluyen a los ligandos de TGF-ß, receptores y moléculas accesorias, activinas y el más grande de estos grupos corresponde a las proteínas morfogenéticas del hueso (BMPs)(21).
Hasta la fecha, se han identificado un 300 variantes diferentes en el gen BMPR2 en el 75% de HAPf, siendo la mayoría variantes “nonsense” que llevan a la haploinsuficiencia(10) y son identificadas mediante el secuenciamiento génico tras la detección de variantes puntuales y en caso de no detectarse alguna variante, se procede a realizar como método complementario el estudio de grandes rearreglos génicos, como el análisis de deleción/duplicación por MLPA (multiplex ligation-dependent probe amplification) u otros métodos similares(5,45) (5, 45), sin embargo, en el 25% restante aun no han sido identificadas las variantes responsables de la enfermedad(20). Respecto a los casos de HAPi, tenemos que se han identificado mutaciones en el gen BMPR2 en el 10–40% de casos(5). Y finalmente respecto a otros genes involucrados en HAP y que también son parte de la vía de señalización de la superfamilia del TGF-B se han identificados mutaciones en ACVRL1 (ALK1), ENG, SMAD4, SMAD9, CAV1, BMPR1B (ALK6)(7,10,32,46-51), lo que enfatiza la importancia de esta via en la integridad de la vasculatura pulmonar.
Diversos estudios muestran que los portadores de variantes en el gen BMPR2 comparado con lo no portadores, presentan una forma mas grave de la enfermedad, con edad de inicio mas temprana y mayor compromiso hemodinámico al momento del diagnóstico y más propensos al trasplante pulmonar(6,9).
El estudio genético-molecular debe ofrecerse a todo paciente con HAPf y HAPi explicándole la importancia de determinar la presencia de variantes germinales en ellos para la posterior búsqueda de la variante específica en otros miembros de su familia en menor tiempo y a menor costo que el primer caso identificado(5). Con el desarrollo de paneles de genes se han disminuido los costos y aumentado la accesibilidad de los pacientes para su realización en comparación con el estudio individual de cada gen.
VIA MOLECULAR DE BMPR2
Los pacientes con HAP portadores de variantes en BMPR2, presentan la enfermedad aproximadamente 10 años antes que los no portadores y tienen un compromiso hemodinámico más severo al diagnostico(9), lo que ha llevado al estudio del gen BMPR2 y la vía molecular por la que actua.
El gen BMPR2 contiene 13 exones(22) y codifica a la proteína BMPR-II de 1038 aminoacidos, que comprende a un dominio extracelular de unión al ligando (exones 2 y 3), un dominio transmembrana (exones 4 y 5), un dominio quinasa catalítico altamente conservado (exones del 6 al 11) y un dominio citoplasmático (exones 12 y 13)(52). Hasta el momento, de las más de 300 variantes diferentes idenficadas, el 30% se localizan en el exon 12(53).
BMPR-II es un receptor de la familia de citosinas conocidas como proteínas morfogeneticas de hueso (BMPs) y como miembro de la superfamilia de receptores TGF-ß, las BMPs juegan un rol crucial en la regulación de la morfogénesis del pulmón(43). Normalmente las BMPs regulan el crecimiento, diferenciacion y apoptosis celular mediante una cascada de señalización intracelular a través de las proteínas citoplasmáticas señalizadoras llamadas Smads. La familia de proteínas Smad son responsables de transformar la señalización del receptor TGF-B y regular la expresión génica(54).
Para entender el mecanismo por el cual las variantes del gen BMPR2 conducen a la HAPh, debe comprenderse la señalización del TGF-B. La señalización a través del BMPRII, al igual que en otros receptores de TGF-B, involucra la unión de la BMP como ligando a un receptor de tipo I (BMPR-I) que se asocia con un receptor de tipo II activo (BMPR-II) en la membrana celular, este BMPR-II fosforila al receptor BMPR-I e inicia una cascada de fosforilaciones sucesivas que involucra a las proteínas Smad que normalmente participan en la inhibición del crecimiento celular e inducción de la apoptosis. De esas proteínas, primero son fosforiladas la Smad 1, 5 y 8, que luego forman un complejo con Smad 4 para luego traslocarse al núcleo para regular la transcripción génica en conjunto con cofactores y represores nucleares específicos(43,55) (figura 1).
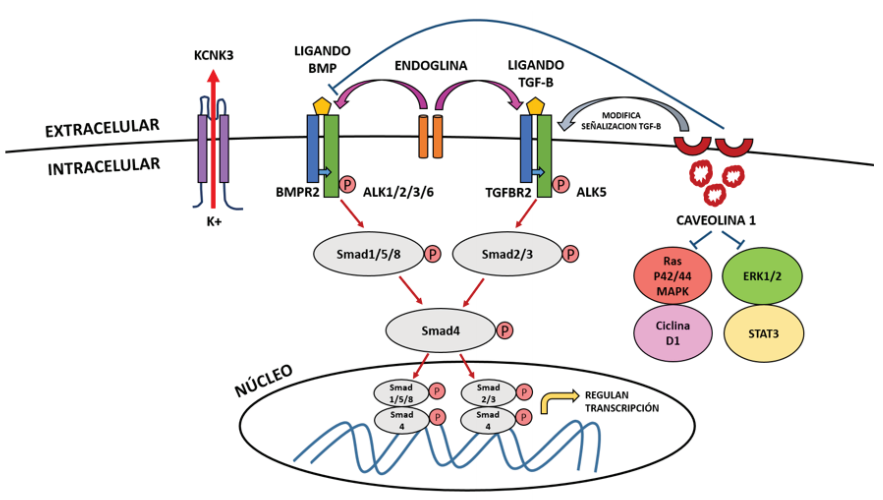
Figura 1. Patogenesis molecular de HAPh
Las variantes en el dominio quinasa de BMPR2 inician una señalización constitutiva cascada abajo a las moléculas Smad en las células musculares lisas de pulmón, causando efectos pro-proliferativos y anti-apoptoticos que promueven el desarrollo de HAP(43), por lo que se postula que estas variantes en el gen BMPR2 eliminan la función regulatoria del crecimiento de las células vasculares pulmonares mediante la disrupción de la activación de Smad.
El ligando BMP se une al receptor tipo II (BMPR2) a nivel de la membrana celular, el cuál fosforila al receptor tipo I (ALK1, ALK 2, ALK 3 o ALK 6) desencadenando la fosforilación de Smad 1, Samd 5, Smad 8, que fosforilan a Smad 4 para luego formar un complejo y traslocarse al nucleo, donde modularan la expresión génica. Este mecanismo es similar a lo que sucede cuando el ligado TGF-B se une al receptor tipo II (TGFBR2), el cuál fosforila al receptor tipo I (ALK 5), desencadenando la fosforilacion de Smad 2 y Smad 3, quienes fosforilan a Smad 4 y se traslocan al núcleo. La endoglina es una glicoproteína accesoria de la membrana que interactua con receptores de señalización para BMP y la superfamilia TGF-B. La caveolina 1 normalmente amortigua la señalización de BMP tras inhibir su receptor para evitar la proliferación vascular, pero en su ausencia causa la activación de las vías de señalización STAT3 y ERK1/2 y de las vías Ras/p42/44/MAPk y Ciclina D1, además modifica la via de señalización de TGF-B proporcionando un vínculo entre las mutaciones de CAV1 y BMPR2 en la patogénesis molecular de HAP. KCNK3 es una proteína de canal de potasio (K+) en las células musculares lisas de las arterias pulmonares y su activación causa salida del K+ al extracelular, hiperpolarizacion de la membrana y vasodilatación.
MECANISMOS DE HAPLOINSUFICIENCIA Y DOMINANTE NEGATIVO
Las variantes heterocigotas en el gen BMPR2 constituyen el principal factor de riesgo para el desarrollo de HAPh, sin embargo los portadores tienen una penentrancia del 20%, lo que ha permitido investigar otros factores que puedan influir en la expresión de la enfermedad(6,40). Las variantes que generan un aminoácido diferente al original pueden ser del tipo “variantes con sentido erróneo” (“missense”) y las “variantes sin sentido” (“nonsense”) que generan una señal de detención de la lectura del ARNm por parte de los ribosomas(43). Aproximadamente el 70% de las variantes patogénicas reportadas en el gen BMPR2 son “nonsense”, “frameshift”, defectos del sitio de “splicing” y rearreglos génicos que llevan a una disminución en su actividad(43) y el 30% restante corresponden a variantes “missense” causando la sustitución en un aminoácido en dominios funcionales importantes del receptor (por ejemplo, unión de ligando, dominio quinasa)(20,56,57).
Las variantes “nonsense” en el gen BMPR2 resultan en una proteína truncada y tienen un impacto diferente a las variantes “missense” debido a que poseen diferencias según la activación de la vía NMD(58). La vía NMD (Nonsense mediated decay), es un mecanismo celular de vigilancia del ARNm por el cual el ARNm truncado producido por una variante “nonsense” es detectado y eliminado por la célula (variantes NMD positivas), impidiendo la traducción de los transcriptos perjudiciales, lo que resulta en un efecto de haploinsuficiencia debido a una insuficiente cantidad de proteína funcional pero con persistencia de la proteina normal remanente producida por el alelo no mutado(59,60).
La haploinsuficiencia es el mecanismo molecular generalmente aceptado para explicar como la expresión del transcripto del alelo no “mutado” del gen BMPR2 influye en la penetrania reducida de la enfermedad(60). En contraste, las variantes “missense” son resistentes a la destrucción por el mecasnimso NMD (variantes NMD negativas), produciendo una proteína mutante con efectos deletéreos que además bloquea completamente la actividad de la proteina remanente del alelo no mutado, creando un efecto llamado “dominante negativo”(22,43,61) que causa una expresión más severa de la enfermedad, con una edad más temprana al diagnóstico y de fallecimiento, así como una menor sobrevida (1,6,60,62-65). (figura 2). En un estudio realizado, se encontró que la mayoría de las variantes “missense” correspondían a pacientes con edad menor de 36 años, mientras que la mayoría de los portadores de variantes “nonsense” tenían una edad de presentación más alla de los 36 años(1).
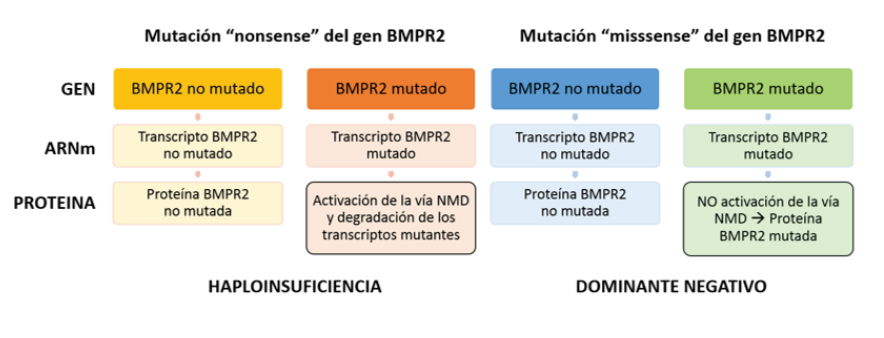
Figura 2. Modelo del Impacto de la Vía NMD
Todos estos hallazgos sugieren que los tipos específicos de variantes nos pueden orientar a diferentes estrategias para nuevas intervenciones(1), es por eso que el tamizaje de BMPR2 en pacientes con HAP es clínicamente útil(66). A pesar de que todos los pacientes HAPf con variantes BMPR2 NMD positivas pueden desarrollar potencialmente la enfermedad, solo el 20% lo hará, es por eso que se postula que la haploinsuficiencia inducida por la variante por si sola no es suficiente para causar HAPf en la mayoría de pacientes y que los niveles de expresión de los transcriptos no “mutados” pueden modificar los grados de haploinsuficiencia(67). Las medidas de tratamiento y prevención dependerán del tipo de variante, como en los casos de variantes “nonsense” donde las terapias que incrementen la expresión de BMPR-II a partir del alelo no mutado pueden ser mas efectivas en prevenir o tratar la enfermedad(6).
La activación de la via NMD resulta en la degradación del transcripto mutante proveniente de una mutación “nonsense”, permaneciendo sólo la proteína BMPR2 normal producida por el alelo no mutado, siendo cuantitativamente reducida pero cualitativamente conservada, lo que conlleva a desarrollar HAP por “haploinsuficiencia”. En cuanto al transcripto mutante resistente a la activación de la via NMD debido a una mutación “missense”, se produce una proteína mutada con función anormal afectando incluso la actividad de la proteína BMPR2 normal producida por el alelo no mutado, donde efecto deletéreo de esta proteína alterada cualitativamente mas no cuantitativamente, resulta en un efecto “dominante negativo” que brinda mayor suceptibilidad a desarrollar HAPf.
OTROS GENES INVOLUCRADOS EN HAP
Se han descrito variantes patogénicas en otros genes además del BMPR2 causantes de enfermedad y que son considerados menos frecuentes (1-3%) (Ver tabla 1). Los genes ACVRL1 (ALK1) y ENG codifican a los receptores ALK1 y endoglina, que son receptores de membrana tipo I de la superfamilia del TGF-B y las variantes heterocigotas en estos genes se asocian directamente a la telangiectasia hemorragica hereditaria (THH), un desorden vascular con herencia autosomico dominante que se cataracteriza por la presencia de telangiectasias cutaneas y malformaciones arteriovenosas que pueden desarrollar HAP(7,10,32,46,51,68-73). Los pacientes portadores de variantes en el gen ALK1 son significativamente más jóvenes al diagnóstico, comparado con los portadores y no portadores de variantes en BMPR2, lo que sugiere una rápida evolución de la enfermedad en estos casos, además de presentar una menor respuesta a la terapia vasodilatadora(7). Las variantes en el gen ALK1 pueden estar presente en niños con HAP a pesar de no tener el cuadro clínico de THH al momento de identificar la variantes, sin embargo, suelen desarrollar la enfermedad posteriormente(32).
Tabla 1. Genes asociados a HAPf
| Tipo de HAP, según MIM | Gen | Localización | Fenotipo MIM | Proteína codificada | Herencia |
| Hipertensión arterial primaria 1 | BMPR2 | 2q33.1q33.2 | 178600 | Receptor de la BMP tipo 2 | AD |
| Hipertensión arterial primaria 2 | SMAD9 | 13q13.3 | 615324 | Proteína relacionada a Sma y Mad 9 | AD |
| Hipertensión arterial primaria 3 | CAV1 | 7q31.2 | 615343 | Caveolina 1 | AD |
| Hipertensión arterial primaria 4 | KCNK3 | 2p23.3 | 615344 | Miembro 3 del canal de potasio, subfamilia K | AD |
| Hipertensión arterial primaria 5, recesivo autosómico | PPHR | No mapeado | 265400 | No identificada | AR |
| Hipertensión arterial asociada a telangiectasia hemorrágica hereditaria 1 | ENG | 9q34.1 | 187300 | Endoglina | AD |
| Hipertensión arterial asociada a telangiectasia hemorrágica hereditaria 2 | ACVL1 | 12q13.13 | 600376 | Receptor de activina 1 A, tipo quinasa II-like. | AD |
| Enfermedad venoclusiva pulmonar tipo 2 | EIF2AK4 | 15q.15.1 | 234810 | Factor de iniciación de la traducción eucariótica, 2-alfa quinasa | AR |
| No clasificado | SMAD1 | 4q31.21 | - | Proteína relacionada a Sma y Mad 1 | AD |
| No clasificado | SMAD5 | 5q31.1 | - | Proteína relacionada a Sma y Mad 5 | AD |
| No clasificado | BMPR1B | 4q22.3 | - | Receptor de la BMP tipo 1B | AD |
| No clasificado | CBLN2 | 18q22.3 | - | Precerebelina | AD |
Las mutaciones en SMAD 1, SMAD 5, SMAD 9 tambien miembros de la familia TGF-B se asocian a HAP y su estudio es solicitado luego que las variantes en BMPR2, ALK1 y ENG han sido analizadas y no se haya encontrado alguna variante(45,47,74).
Otro gen involucrado en HAP es el gen CAV1 que regula la fosforilacion de Smad2 y Smad3, y las variantes en este gen son causa rara de HAP(10,48). El gen CAV1 codifica a una proteína llamada caveolina-1, necesaria para la formación de caveola, que es crucial para la unión de los receptores de membrana y la iniciación de la cascada de señalización celular con la via TGF-ß. Por otra parte, esta vía controla la señalización del crecimiento, diferenciación y apoptosis de diversos tipos de células como las células endoteliales vasculares pulmonares (ECs) y del músculo liso (SMCs)(19).
El gen KCNK3 es un miembro de los canales de potasio con dominio de dos poros expresado en las células del musculo liso de las arterias pulmonares, y las variantes en el gen KCNK3 son una rara causa de HAPf y HAPi(75).
El gen CBLN2 codifica a la cerebellin 2, que se expresa en el pulmón y ha sido encontrado principalmente en pulmones explantados de pacientes con HAP y en las células endoteliales cultivadas de los mismos, por lo que se ha planteado que actúen en la proliferación celular(76). Algunos estudios han revelado que las variantes en el gen de la precerebelina 2 (CBLN2) pueden incrementar el riesgo de HAP en casi el doble comparado a las variantes en el gen BMPR2(10).
Otros genes recientemente reconocidos como causas posibles de HAP son los genes SMAD9 y BMPR1B (ALK6), encontrados principalmente en casos de HAPi(47,49).
Variantes en el gen EIF2AK4 han sido identificados en multiples familias con hemangiomatosis capilar pulmonar (PCH)/HAP asociada a enfermedad venooclusiva (PvOD) siendo variantes homocigotas o heterocigotas compuestas en el gen EIF1AK4, correspondiente a una herencia autosómica recesiva(77). El producto proteico de EIF2AK4 pertenece a la familia de quinasas donde la subunidad alfa de esta proteína juega un rol crítico en la inducción de la angiogénesis, proliferancion y resistencia a la apoptosis en estados de estrés celular, además se ha encontrado que la proteína eIF2aK4 interactua con SMAD4, SMAD1, ALK-1, ENG y TGFBR2, similar a la vía molecular del gen BMPR2(78).
Actualmente los métodos de tamizaje genético estándar se enfocan en el secuenciamiento de las regiones exónicas codificantes de los genes BMPR2, ACVL1, ENG, SMAD9, SMAD1 Y SMAD5 que están involucradas en la vía de señalización de TGF-ß(19). Los estudios genéticos han permitido el mejor entendimiento de las bases moleculares de la HAP, sin embargo, cerca de 30% de los casos de HAPh y 60-90% de los casos de HAPi no poseen variantes en los genes BMPR2, ALK1, ENG ni SMAD9, por lo que se plantea que existan otros genes en la superfamilia de TGF-ß u otras vías de señalización como BMP/MAPk, p38, Toll-like y vía Rho quinasa entre otras, que puedan estar asociadas con el desarrollo de HAPi y HAPh(79). Las regiones intrónicas, las cuáles aun no han sido del todo estudiadas, son el siguiente paso a investigar, debido a que se plantea la hipótesis que también existan variantes a ese nivel(80), donde el secuenciamiento de próxima generación nos permite realizar un análisis rápido y completo de cada paciente.
ASESORIA GENÉTICA EN HAP
La asesoría genética es el proceso por el cual se provee al paciente y a sus familiares la información necesaria sobre las causas, herencia e implicancias del desorden genético que posee, mediante la recolección de datos de la historia familiar del paciente, elaboración del heredograma y solicitud de estudios genéticos-moleculares, siendo estos últimos lo que nos permiten determinar el estado “mutacional” de los miembros de una familia.
Para establecer el diagnóstico de HAPh, es necesario confirmar el diagnóstico en al menos dos familiares (HAPf) o identificar la variante germinal en un caso aislado en la familia (HAPi); sin embargo, existen factores que llevan a no reconocer los casos de HAPh como la penetrancia incompleta, una inadecuada historia familiar, un diagnóstico incorrecto de otros miembros afectados en la familia o la imposibilidad de recurrir a estudios moleculares que detecten variantes como BMPR2(3).
La HAPh (MIM #178600)(53), es definida como un trastorno genético, con patrón de herencia autosómico dominante (AD), debido a la presencia de una mutación germinal heterocigota principalmente en el gen BMPR2, con penetrancia incompleta del 20%(3), edad de presentación variable y predominancia en mujeres(58). Por ser un trastorno AD, cuando un paciente porta mutación en el gen BMPR2 posee un riesgo de trasmitirlo a su descendencia en un 50%, pero debido a que posee una penetrancia del 20%, el riesgo a desarrollar HAP sería 10% (50% x ~20%).
La penetrancia incompleta y la expresividad variable son las mayores dificultades para la asesoría genética, debido a que pueden haber casos “de novo” que son aislados en la familia (HAPi) y porten una variante germinal por lo que deben ser informados sobre los estudios genéticos-moleculares disponibles(17) o podrían existir varios miembros de la familia portadores de la variante que no han desarrollado aún la enfermedad y puede solapar una historia familiar positiva y que al momento de la elaboración del heredograma podrían no ser identificados(17,81-84).
Los estudios genéticos-moleculares deben solicitarse en los casos de HAPf y HAPi como parte del estudio de la enfermedad(1), pero debemos tener en cuenta que previo al estudio genético y posterior a su resultado debe brindarse la asesoría genética al paciente y a sus familiares(5), ya que se deberá explicar al paciente los resultados que se obtendrán, como los resultados “positivos”, donde se identifica a la variantes causal en el gen analizado; resultados “negativos”, donde no se ha identificado la variante causal en el gen analizado, resultados con “variantes de significado incierto” en los que la variación génica encontrada no ha sido reportada previamente como causal de enfermedad y debería esperarse otros resultados similares asociados a enfermedad para ser considerada patogénica; y finalmente los resultados “verdaderamente positivos” y “verdaderamente negativos” en los que tras la identificación de una variante causal de un determinado gen en el paciente afectado se procede a la búsqueda especifica de la misma mutación en otros miembros de la familia, si resulta positiva esto indicará que ha heredado el gen causal de la enfermedad y si el resultado fuese negativo indicaría que el familiar no ha heredado la variante germinal(83).
Debemos tener en cuenta que la realización de estos estudios genéticos-moleculares y los resultados obtenidos, pueden tener implicancias psicológicas en el paciente y sus familiares, por lo que se debe tener en cuenta que el resultado se les dará a conocer a los pacientes afectados y familiares en riesgo que deseen conocerlo(17), además que el manejo del paciente con HAP deben ser realizado siempre por un equipo mutidisciplinario con médicos especialistas en HAP, médicos genetistas, asesores genéticos, psicólogos y enfermeras principalmente, para el manejo integral del paciente y familiares(17,84). Por las implicancias que pueden tener los resultados moleculares en todos los pacientes con trastornos genéticos, en Mayo del 2008 en EEUU, se creó el GINA (Genetic Information Nondiscrimination Act), que protege a los miembros de discrimacion en el trabajo en base a su predisposición genética(5).
Una vez obtenido el resultado del estudio genético-molecular en el paciente, es importante ofrecer las medidas de seguimiento correspondientes, como es el caso de los portadores de una variante en el gen BMPR2 a quienes se les debe solicitar ecocardiografías de control y realizar la asesoría genética correspondiente, de este modo si es que se presenta la enfermedad se pueda realizar un diagnóstico temprano y brindar una terapia oportuna(43),83-86) (43, 83-86), esto ha sido recomendadas en diversas guías de hipertensión pulmonar. Con relación a lo anterior, mucho se discute sobre si existe verdaderamente el fenómeno de anticipación o es que posiblemente los familiares portadores asintomáticos se encuentran alertas a la enfermedad y se realiza un diagnóstico más temprano de la enfermedad(3).
Aún quedan muchas variantes génicas por descubrir en todas las formas de HAP, para lo cual actualmente contamos con tecnología como el secuenciamiento de próxima generación que nos facilitan el descubrimiento de variantes poco frecuentes en el estudio del genoma completo del paciente.
CONCLUSION
La HAP es una enfermedad heterogénea compleja y la identificación de los genes causales en los pacientes afectados con HAPf es relevante para la asesoría genética y para realizar la búsqueda de la variante en el resto de sus familiares asintomáticos en riesgo con un fin predictivo. La adecuada recopilación de los datos de la historia familiar del paciente para referirlo para la asesoría genética correspondiente y poder brindarle las opciones de seguimiento y terapéuticas en relación a los resultados moleculares de cada paciente.
Contribuciones de Autoría: Los autores participaron en la génesis de la idea, diseño de proyecto, recolección e interpretación de datos, análisis de resultados y preparación del manuscrito del presente trabajo de investigación.
Financiamiento: Autofinanciado.
Conflictos de intereses: Los autores declaran no tener conflicto de interés.
Recibido: 16 de abril 2020
Aprobado: 31 de mayo2020
Correspondencia: Maria del Carmen Castro Mujica
Dirección: Av. Alfredo Benavides 5440, Santiago de Surco
Teléfono: 987597107
Correo: mc.castro.mujica@gmail.com
REFERENCIAS BIBLIOGRÁFICAS
