ARTICULO DE REVISIÓN
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2022 - Universidad Ricardo Palma10.25176/RFMH.v22i3.5043
IMPLICANCIAS CLÍNICAS DE LA BIOLOGÍA MOLECULAR DEL CÁNCER DE PRÓSTATA: ARTÍCULO DE REVISIÓN
CLINICAL IMPLICATIONS OF THE MOLECULAR BIOLOGY OF PROSTATE CANCER: REVIEW ARTICLE
María del Carmen Castro-Mujica1,a
1Facultad de Medicina Humana, Universidad Ricardo Palma, Lima-Perú.
a Médico genetista.
RESUMEN
Para enteder el término de heterogeneidad genómica en cáncer de próstata debemos comprender la evolución genómica clonal del cáncer, así como saber que es un fenómeno dinámico y evolutivo. Conocer el genoma del cáncer de próstata no solo nos permite tener una visión en el tiempo de las alteraciones genómicas que se producen durante sus diferentes estadíos, sino también conocer sobre los mecanismos de la metástasis. Además, conocer el componente hereditario del cáncer de próstata permite la evaluación de los pacientes y poder identificar si estamos frente a una familia en riesgo.
Palabras Clave: Neoplasias de la Próstata; Heterogeneidad Genética; Mutación de Línea Germinal.(fuente: DeCS BIREME).
ABSTRACT
To understand the term genomic heterogeneity in prostate cancer, we must understand the clonal genomic evolution of cancer, as well as knowing that it is a dynamic and evolutionary phenomenon. Knowing the genome of prostate cancer not only allows us to have a vision over time of the genomic alterations that occur during its different stages, but also to learn about the mechanisms of metastasis. In addition, knowing the hereditary component of prostate cancer allows the evaluation of patients and to be able to identify if we are dealing with a family at risk.
Keywords: Prostatic Neoplasms; Genetic Heterogeneity; Germ-Line Mutation. (Source: MeSH NLM).
INTRODUCCIÓN
El cáncer de próstata es, desde el punto de vista molecular, biológicamente heterogéneo debido a la diversidad de alteraciones moleculares inter e intratumorales, debido a que es parte de un proceso genómico dinámico y evolutivo. Con el desarrollo de nuevas técnicas moleculares, como la secuenciación génica, micromatrices, estudios epigenenómicos, entre otros, se ha podido caracterizar molecularmente al cáncer de próstata, encontrándose diferencias incluso entre los diferentes estadíos de la enfermedad.
El cáncer de próstata es, desde el punto de vista molecular, biológicamente heterogéneo debido a la diversidad de alteraciones moleculares inter e intratumorales, debido a que es parte de un proceso genómico dinámico y evolutivo. Con el desarrollo de nuevas técnicas moleculares, como la secuenciación génica, micromatrices, estudios epigenenómicos, entre otros, se ha podido caracterizar molecularmente al cáncer de próstata, encontrándose diferencias incluso entre los diferentes estadíos de la enfermedad.
El objetivo de este artículo de revisión es presentar las alteraciones moleculares a nivel somático en el cáncer de próstata, su clasificación molecular, heterogeneidad genómica, evolución clonal, biomarcadores, así como los síndromes de predisposición genética al cáncer de próstata.
ALTERACIONES MOLECULARES A NIVEL SOMÁTICO
Las principales alteraciones moleculares descritas en el cáncer de próstata incluyen a la fusión génica TMPRSS2-ETS, variantes en el número de copias de los genes TP53, AR, RB1, PTEN/PIK3CA, BRCA2 y ATM, entre otras.
TMPRSS2 (Transmembrane protease, serine 2) es una serina proteasa regulada por andrógenos. ETS (erythroblastosis virus E26 oncogene homolog) es una de las familias más grandes de factores de transcripción que incluye al ERG y ETV1, los cuáles se fusionan con el gen TMPRSS2 en aproximadamente el 50-79% de los casos de cáncer de próstata 1. Esta fusión se debe a la deleción de una región entre ambos genes y se asocia a un peor pronóstico en los casos localizados 2.
La vía de señalización PTEN/PI3K/AKT juega un rol primordial en la regulación del crecimiento y muerte celular, mientras que la vía PI3K/AKT/mTOR juega un rol fundamental en la metástasis tumoral 3. Las variantes en los genes PTEN y PI3K son mutuamente excluyentes ya que el gen PTEN es un regulador negativo de la vía PI3K/AKT por lo que la pérdida de PTEN se asocia a un peor pronóstico 1. El 2-14% de los casos de cáncer de próstata poseen variantes en el gen PTEN y el 12-41% de casos poseen pérdida en el número de copias 4. Por otro lado, en el 3-4% de casos de cáncer de próstata se han descrito variantes en el gen PI3K y su amplificación se ha reportado en 4-10% de casos y además se han descrito casos de variantes combinadas de PTEN y TP53, los cuáles son muy agresivos 4.
El gen TP53 es un supresor tumoral que juega un rol muy importante en el mantenimiento de la estabilidad genómica y previniendo la carcinogénesis. Aproximadamente el 3-47% de los casos de cáncer de próstata poseen variantes en el gen TP53 y entre 2-15% pérdida del gen 4. Variantes en el gen TP53 se han asociado a un mayor riesgo de recurrencia de la enfermedad 1.
Alrededor del 2-18% de los casos de cáncer de próstata poseen variantes en el gen del receptor de andrógenos AR (androgen receptor gen) o una amplificación génica (5-52% de casos), siendo muy frecuente en los casos refractarios a terapia hormonal 4. Los receptores de andrógenos (AR) pertenecen al grupo de receptores nucleares de hormonas esteroideas y actúa como un factor de transcripción ligando-dependiente que controla la expresión de genes específicos 1.
La proteína RB es producto del gen RB1 (retinoblastoma 1), un supresor tumoral el cuál está mutado en aproximadamente el 1-4% de casos de cáncer de próstata, mientras que en el 5-23% se ha demostrado pérdida del gen RB1 4. Una alteración en el gen RB1 conlleva a una falla en la función de la proteína estimulando la salida del receptor de andrógenos y conferiendo resistencia a la castración 1. El gen RB1 está frecuentemente delecionado o metilado en el cáncer de próstata resistente a castración 1.
El gen APC es un supresor tumoral que codifica una proteína que actua de forma antagónica a la via de señalización Wnt 1. La hipermetilación del promotor del gen APC es un predictor de mal pronóstico en cáncer de próstata. Aproximadamente el 3-10% de los casos de cáncer de próstata poseen variantes en el gen APC 4.
El protooncogen MYC codifica a una proteína que juega un rol importante en la progresión del ciclo celular, apoptosis y transformación 1. La activación de este protooncogen lo transforma en un oncogen que se expresa mediante amplificación génica, lo que estimula el desarrollo del cáncer, que se encuentra en aproximadamente el 2-20% de casos de cáncer de próstata 4.
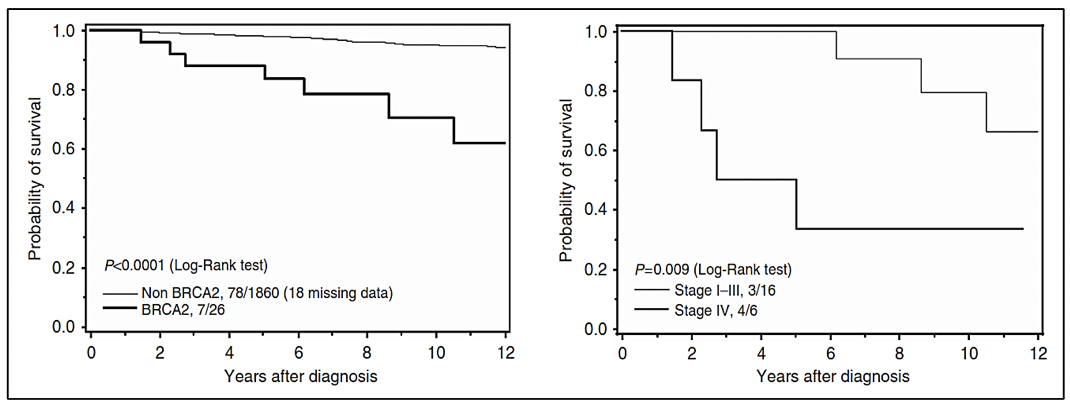
BRCA2 es un gen supresor de tumores, que se encarga de mantener la estabilidad genómica principalmente en la vía de la reparación del ADN de doble cadena por recombinación homóloga 1. Aproximadamente el 9% de los casos de cancer de próstata presentan variantes en BRCA2, de los cuales el 2-6% son debido a variantes germinales, las cuales son un factor pronóstico de sobrevida en todos los estadíos del cáncer de próstata incluyendo los casos localizados 5. Cuando las variantes son germinales, los familiares del paciente poseen una mayor probabilidad de haber heredado el gen mutado, confiriéndoles un riesgo a desarrollar alguna neoplasia relacionada al síndrome de cáncer de mama/ovario hereditario en los portadores.
ATM es un gen reparador del ADN que actúa a nivel del ciclo celular como un controlador necesario para la respuesta celular al daño del ADN y para mantener la estabilidad genómica 1. Aproximadamente el 5% de los casos de cáncer de próstata presentan variantes somáticas en ATM, incluyendo 1% de variantes germinales 5.
FUSIÓN GÉNICA TMPRSS2-ETS
En el años 2005, Scott Tomlims y colaboradores identificaron un rearreglo recurrente en más de la mitad de los casos de cáncer de próstata analizados, el cual conllevaba a la fusión génica del gen TMPRSS2 (21q22) con miembros de la familia de factores de transcripción ETS (ERG, ETV1 y ETV4 localizados en los locus 21q22, 7p21 y 17q21, respectivamente) (6-8). Evaluar la presencia de la fusión génica TMPRSS2-ETS en las muestras de cáncer de próstata sirve como marcador pronóstico de la enfermedad. La determinación de la sobreexpresión de ERG por inmunohistoquímica ha sido altamente correlacionada con el estatus de la fusión génica con un 86% de sensibilidad y especificidad 6.
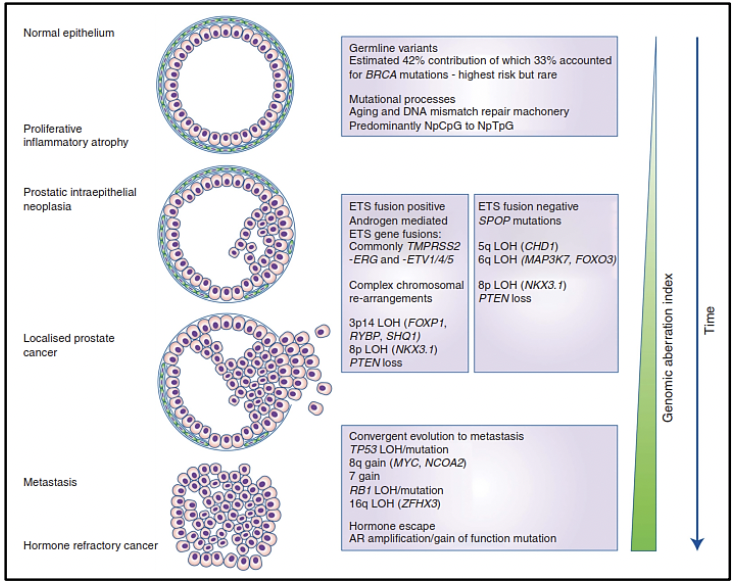
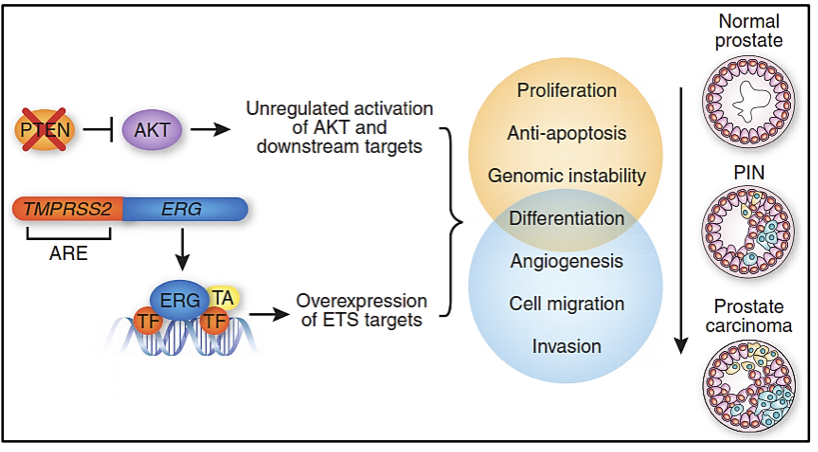
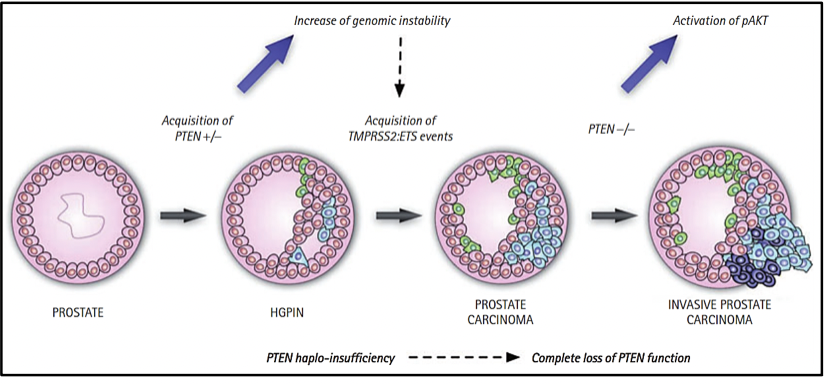
CLASIFICACIÓN MOLECULAR DEL CÁNCER DE PRÓSTATA
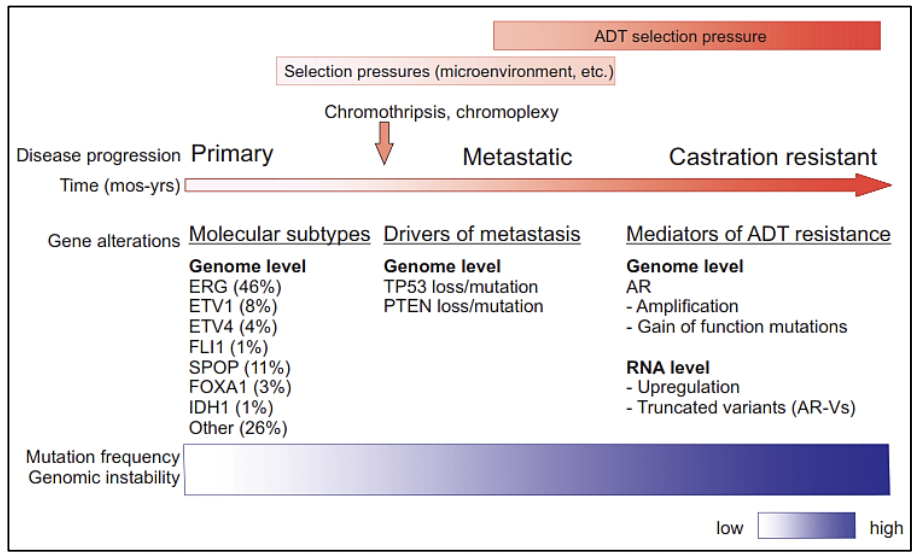
Actualmente existe una clasificación molecular del cáncer de próstata primario. Los siete subtipos moleculares del cáncer de próstata primario, considerados conductores tempranos de la carcinogénesis, están definidos por las fusiones ERG (46%), fusiones o sobreexpresión de ETV1/ETV4/FLI1 (8%, 4%, 1%, respectivamente), o por variantes en los genes SPOP (11 %), FOXA1 (3%) e IDH1 (1%) (9-11), mientras que un 26% que está compuesto por otras variantes. Así mismo, se ha determinado que el perfil molecular en el cáncer de próstata primario y metastásico son diferentes.
HETEROGENEIDAD GENÓMICA EN CÁNCER DE PRÓSTATA
Para enteder el término de heterogeneidad genómica en cáncer de próstata debemos comprender la evolución genómica clonal del cáncer así como saber que es un fenómeno dinámico y evolutivo. La heterogeneidad puede ser intratumoral e intertumoral, asi como en los diferentes estadíos del cáncer de próstata, es por eso que actualmente hablamos subtipos moleculares en cáncer de próstata. La alta heterogeneidad tumoral tiene implicancias en el diagnóstico, seguimiento y tratamiento de los pacientes con cáncer de próstata.(12)
Estudios moleculares como la secuenciación génica genómico de próxima generación, así como estudios de células tumorales circulantes, estudios epigenéticos, micromatrices, entre otros, nos proporcionan la evidencia de una expansión clonal del cáncer de próstata con los diferentes eventos mutacionales que se producen en su genoma a nivel somático en diversas etapas de la progresión de la enfermedad. Existen investigaciones de la composición genómica de múltiples sitios de metástasis en un mismo paciente o han seguido la evolución clonal longitudinalmente en los tejidos. Para comprender el origen del cáncer de próstata, su comportamiento en el tiempo y los riesgos de progresión de enfermedad es necesario conocer sobre la evolución genómica espacial y temporal del cáncer de próstata.
La arquitectura clonal de los tumores primarios y metastásicos nos permiten conocer la evolución clonal
durante la progresión de la enfermedad 13. Diversas variantes así como vías
moleculares alteradas tanto en el tumor primario como en la metástasis han sido identificadas,
confirmando la heterogeneidad intratumoral y evidenciando así que la metástasis y la resistencia al
tratamiento por deprivación de andrógenos ocurren debido a diferentes alteraciones moleculares
adquiridas en el tiempo 13.
Estudios revelan que la pérdida o variante del gen TP53 así como la pérdida de PTEN ocurren antes o al
principio de la metástasis, lo que indica que son conductores de la diseminación metastásica 13. Además de los 7 subtipos moleculares ya mencionados (fusiones ETS,
variantes en FOXA1, FLI1, SPOP e IDH1) puede existir una adquisición de nuevas variantes que conducen
a las metástasis, las cuáles podrían no presentarse preferentemente en alguno de estos subtipos 13.
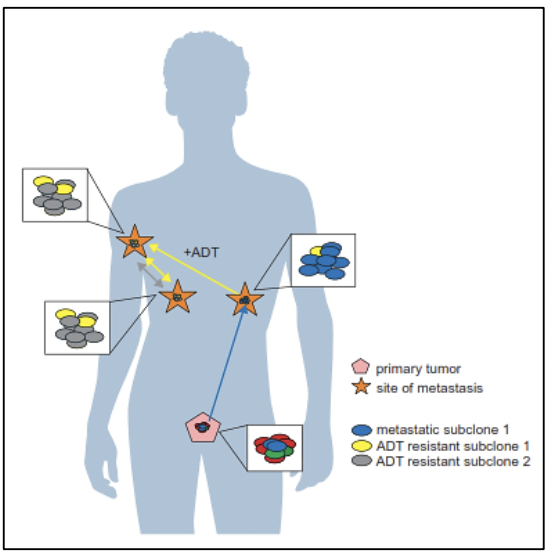
Otros hallazgos importantes como el del receptor de andrógenos (AR) el cuál se encuentra alterado en más del 60% de casos de cáncer de próstata metastásico y el cual es también un mediador para la resistencia a la terapia por deprivación de andrógenos (ADT) puede adquirir nuevas variantes luego de producida la metástasis 14. Lo que aun no se conoce con claridad es que si estas subclonas raras originadas en el tumor primario o tempranamente en las metástasis, poseen alteraciones en AR que posteriormente promueven la resistencia a la ADT o si es que surgen estas alteraciones después de la metástasis y el tratamiento inicial con ADT 13.
El estudio del genoma del cáncer de próstata no solo nos permite tener una visión en el tiempo de las alteraciones genómicas que se producen durante los diferentes estadíos del cáncer de próstata, sino también conocer sobre los mecanismos de la metástasis. La diseminación metastásica puede ocurrir a través de la siembre monoclonal o policlonal entre metástasis o en ondas que se originan del tumor primario 14. Se ha demostrado además que la propagación clonal no es sólo unidireccional, por ejemplo cuando las subclonas metástasicas llegan al lecho quirúrgico del tumor primario resecado.Figura 1 y Figura 2
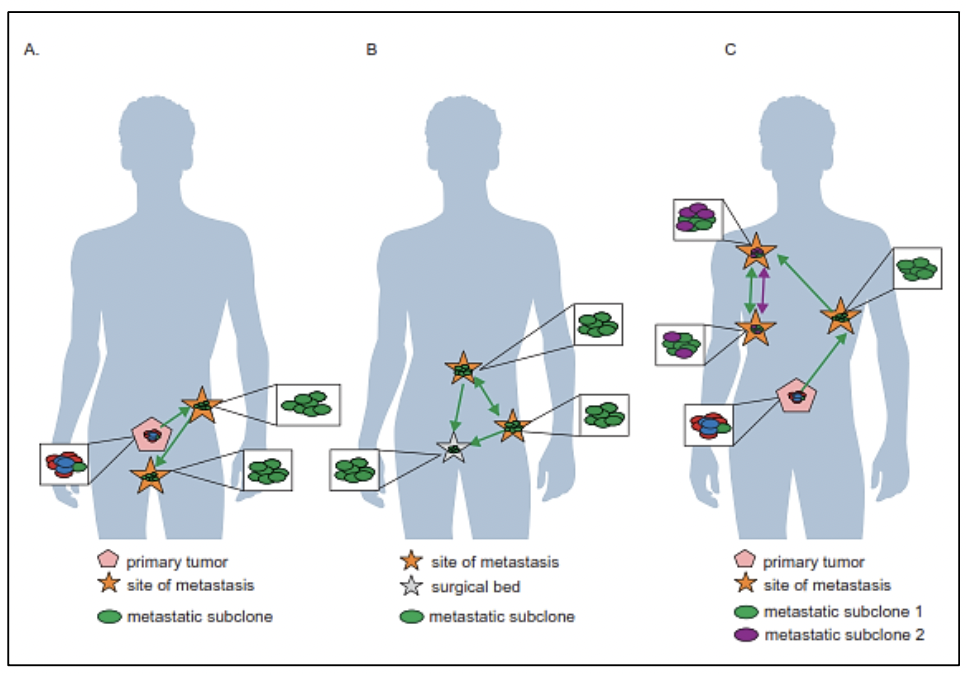
Existe evidencia que tras la prostatectomía radical, las piezas tumorales poseen múltiples focos de cáncer intraprostáticos los cuales están separados y pueden ser biológicamente variables en su potencial de agresividad y progresión de enfermedad 15. La heterogeneidad puede ser de dos tipos, intratumoral (diferentes alteraciones moleculares en los múltiples focos de cáncer en la pieza tumoral de un mismo paciente) así como intertumoral (como por ejemplo, diferentes alteraciones moleculares en pacientes con mismo score de Gleason) 15. Mediante la secuenciación génica de próxima generación, se han podido identificar una diversidad de alteraciones moleculares en el cáncer de próstata como fusiones génicas, amplificaciones, deleciones hemicigotas y homocigotas y/o variantes puntuales, lo que muestra que existe una heterogeneidad genómica.
El cáncer de próstata, al originarse mediante un proceso evolutivo clonal debido a un acumulo secuencial de variantes hasta el punto en que estas variantes promueven el desarrollo de un fenotipo neoplásico (estas variantes son llamadas “drivers” o conductoras) 16, difiere en su composición molecular en cada estadío de la enfermedad. Algunas de estas variantes conductoras aceleran la adquisición de nuevas variantes al obstaculizar la actividad de algunos mecanismos celulares para detectar o reparar el daño del ADN, o para responder con muerte celular programada. El aumento de la tasa de mutación lleva a la adquisición de variantes que no tienen relevancia funcional inmediata para la célula (variantes pasajeras) pero que más adelante tendrán relevancia o incluse ser vulnerables como blancos terapéuticos (17-19).
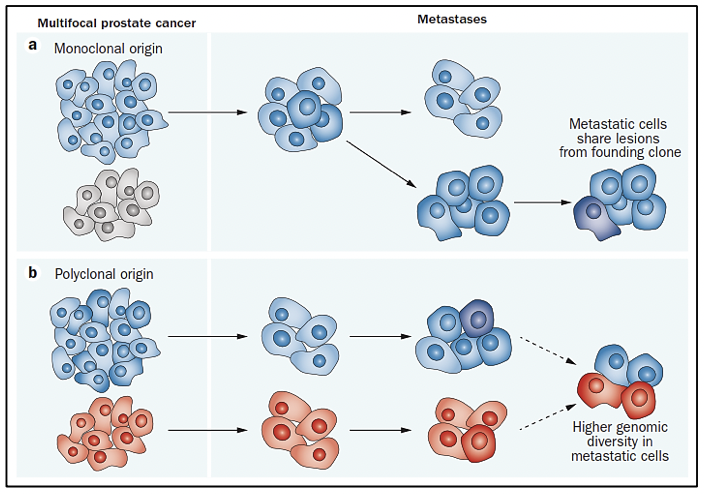
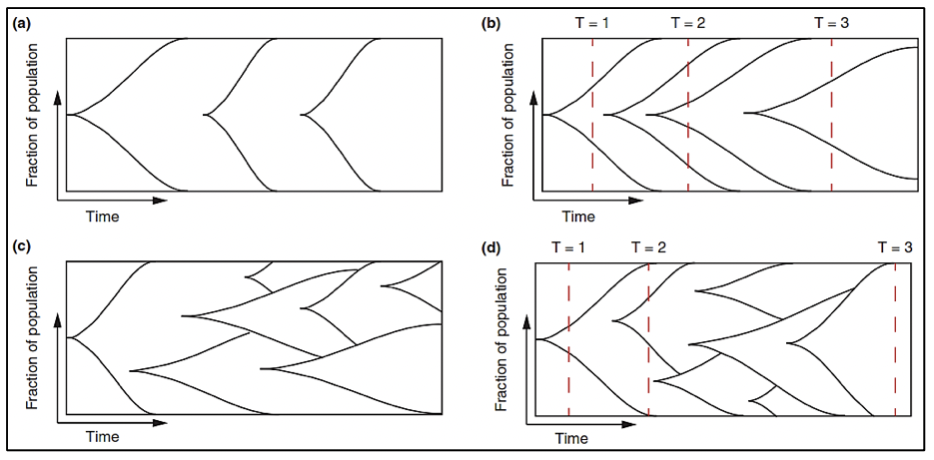
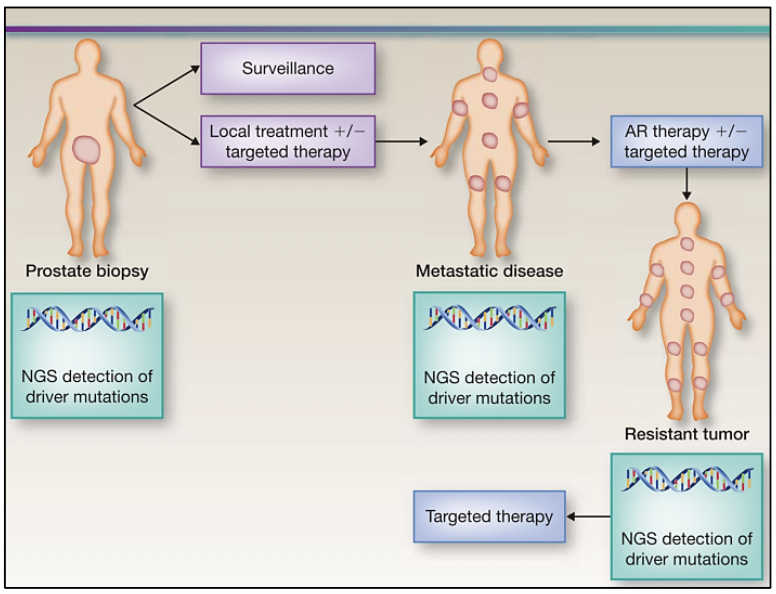
Para realizar una medicina de precisión en pacientes con cáncer de próstata es necesario que al momento de realizar el diagnóstico mediante una biopsia, se solicite el análisis molecular para la detección de variantes conductoras en el tumor y en base al perfil molecular que posea, determinar el seguimiento y tratamiento. Posteriormente, si la enfermedad progresa o si existe resistencia al tratamiento es necesario también realizar un análisis molecular, debido a que el perfil molecular es variable en cada estadío de la enfermedad.
BIOMARCADORES EN CÁNCER DE PRÓSTATA
Los biomarcadores son muy utilizados para el tamizaje, detección y pronóstico del cáncer de próstata, revolucionando el diagnóstico y seguimiento de la enfermedad. A pesar de diversos estudios en cáncer de próstata, no existe un biomarcador molecular con potencial aplicación clínica para el diagnóstico y la estratificación en todos los estadíos de la enfermedad, desde la localizada hasta la metastásica. Usualmente se utiliza el antígeno prostático específico (PSA) de forma rutinaria como un marcador, sin embargo tiene un bajo valor predictivo positivo en el cáncer de próstata localizado y no permite diferenciar el estadío de la enfermedad (20-21).
El PCA3 (9q21-22) es un mRNA no codificante altamente expresado en tumores prostáticos, el cuál puede ser detectado en orina y en fluido prostático en pacientes con cáncer de próstata, además de ser aprobado por la FDA y estar disponible en diversos laboratorios en el mundo. El punto de corte aprobado por la FDA es de 25, ya que es en ese punto donde existe un equilibrio entre la sensibilidad y especificidad de la prueba 22. Existen varios estudios que también utilizan el punto de corte 35 22. Un valor de PCA3 > 35 en orina tiene una sensibilidad de 66% y especificidad de 76% para el diagnóstico de cáncer de próstata comparado con el PSA en suero (especificidad 47% y sensibilidad 65%) 23. El estudio de PCA3 en orina obtenido luego de un masaje prostático es superior al dosaje de PSA en suero en predecir el resultado de la biopsia con una sensibilidad y especificidad de 70% y 80% respectivamente, además posee un valor predictivo negativo de 90% 23. El examen en orina puede predecir la presencia del cáncer de próstata en una biopsia, siendo los los niveles de PCA3 independientes al volumen prostático y al PSA en suero 23. Incluso se ha demostrado elevados niveles de PCA3 en pacientes con PSA elevado y con biopsias negativas, lo que podría ayudar a reducir el número de biopsias innecesarias 23.
Además de ser un rearreglo prevalente en el cáncer de próstata, la fusión génica TMPRSS2-ERG sirve como un biomarcador molecular en cáncer de próstata con una especificidad de 90% y un valor predicitivo positivo del 94% 24. Con la detección de esta fusión génica, la probabilidad de encontrar cáncer en la biopsia aumenta de un 15% a un 90%, e incluso si se detecta la fusión génica en orina y tras la biopsia no se detecta cáncer, es necesario repetir la biopsia debido a la alta especificidad de este biomarcador 24. Además, esta fusión génica se asocia a un mal pronóstico en los pacientes que la porten en el tumor 23. La combinación de la detección de TMPRSS2-ERG y PCA3 en orina, mejora el rendimiento de sólo el uso del PSA para la detección del cáncer de próstata y la predicción clínicamente significativa del cáncer 25.
Las alteraciones genéticas a nivel germinal son biomarcadores moleculares que permiten el cálculo de riesgo en pacientes con cáncer de próstata, debido a que se ha demostrado que influye en la agresividad del cáncer de próstata y en la sobrevida de los pacientes 23. Además, la presencia de variantes en algunos de estos genes, se asocia a la presentación más temprana del cáncer así como antecedentes familiares con cáncer de próstata.
Otros biomarcadores prometedores en cáncer de próstata son los cambios en la metilación del ADN, acetilación de histonas o los microRNAs, los cuales pueden llevar al silenciamiento génico resultando en una alteración en la expresión génica sin alterar la secuencia del ADN 23. La hipermetilación del gen glutatión S-transferasa P1 (GSTP1), es uno de los prevalentes (90%), y puede ser detectada tanto en suero como orina de pacientes, sin embargo no es específica del cáncer de próstata ya que se ha visto en aproximadamente el 70% de casos de neoplasia intraepitelial de alto grado 23.
ALTERACIONES EPIGENÉTICAS Y ESTUDIOS DE METILACIÓN EN CÁNCER DE PRÓSTATA
Los cambios en la metilación del ADN junto con el silenciamiento epigenético de algunos genes son los cambios somáticos más tempranos identificados en el desarrollo del cáncer de próstata 26. El gen GSTP1 codifica una enzima responsable de proteger a la célula de los daños en el genoma 6. La pérdida de expresión de GSTP1 es un evento temprano en el inicio de la carcinogénesis, ya que se ha demostrado metilación del gen GSTP1 en 5-10% de los casos de atrofia proliferativa inflamatoria y en más del 70% de las neoplasias prostáticas intraepiteliales de alto grado 6. La metilación del promotor GSTP1 se asocia a un riesgo de recurrencia en pacientes con cáncer de próstata, lo cual lo convierte en un marcador de recurrencia 6.
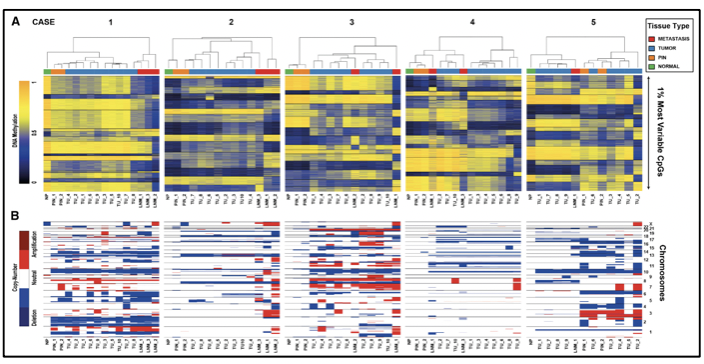
CÉLULAS CIRCULANTES TUMORALES EN CÁNCER DE PRÓSTATA
Entre los nuevos biomarcadores moleculares utilizados en el cáncer de próstata, se encuentra la detección de células tumorales circulantes (CTC) en la sangre periférica y es utilizada como una herramienta de pronóstico ya que se ha propuesto que la propagación de las CTC en la sangre es un mecanismo esencial de las metástasis 21. La detección de CTCs está aprobada por la FDA para el seguimiento del tratamiento del cáncer de próstata metastásico 21. En el cáncer de próstata metastásico, el umbral de CTC por 7,5 ml de sangre venosa se ha determinado como marcador pronóstico de sobrevida global de forma significativa 21.
PREDISPOSICIÓN GENÉTICA AL CÁNCER DE PRÓSTATA
El cáncer de próstata posee un componente hereditario, el cual se caracteriza por la presencia de cáncer a temprana edad, poseer antecedentes familiares de cáncer de próstata y otros tumores, debido a variantes germinales en genes BRCA1/2, MMR (MLH1, MSH2, MSH6, PMS2), HOXB13, entre otros 27.
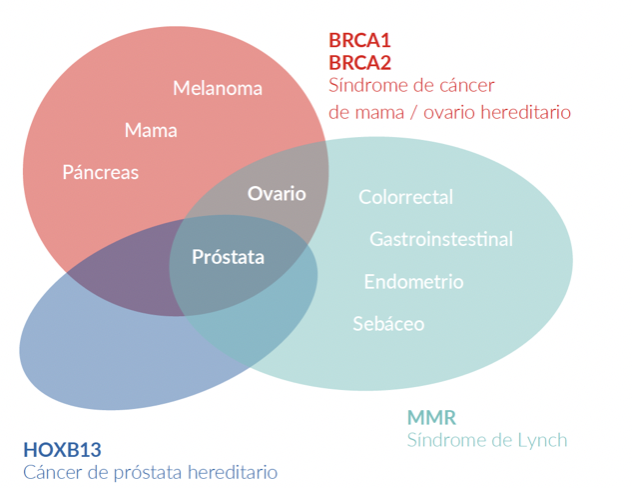
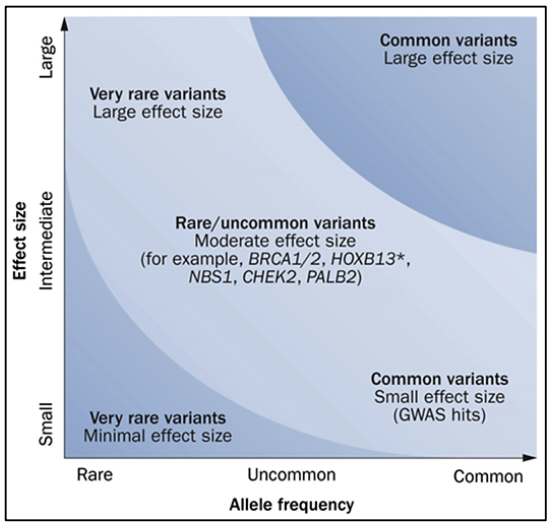
Criterios de referencia para evaluación de genética de pacientes con cáncer de próstata 27:
Para la evaluación de pacientes con sospecha de predisposición genética al cáncer de próstata, es necesario realizar un árbol genealógico para poder identificar si estamos frente a una familia en riesgo. Según los tipos de neoplasias, edad de presentación y patrón de herencia, podremos definir el síndrome genético y así dirigir el estudio molecular a fin de identificar variantes germinales en el paciente y en sus familiares.
El gen HOXB13 ha sido identificado como un gen de susceptibilidad a cáncer de próstata 28. Las variantes germinales en el gen HOXB13 se han asociado a un incremento del riesgo en 2 a 8 veces, siendo estos casos los correspondientes al Cáncer de próstata hereditario 27. Una variante germinal en HOXB13, la cuál es recurrente, G84E, p.(Gly84Glu), c.251G>A, ha sido reportada frecuentemente en pacientes con diagnóstico de cáncer de próstata a edad temprana (2.2%) e historia familiar (3.1%) 28.
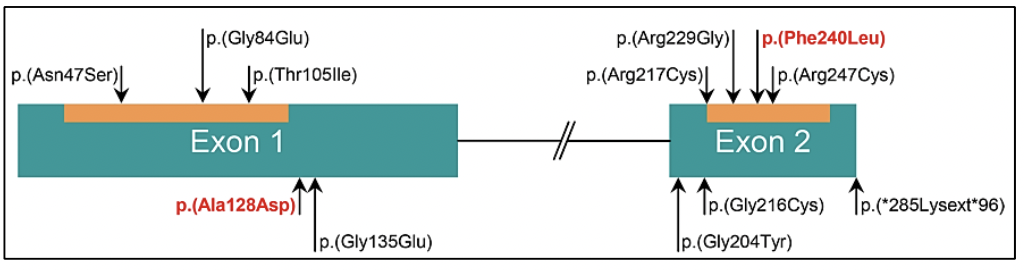
Las variantes germinales en los genes BRCA1 y BRCA2 corresponden a los casos de síndrome de cáncer de mama ovario hereditario, donde además del riesgo de cáncer de mama/ovario, melanoma, páncreas, existe un riesgo en los varones a desarrollar cáncer de próstata 27. Las variantes germinales en el gen BRCA2 se han viso asociadas a un fenotipo más agresivo de la enfermedad 29. Así mismo, las variantes germinales en el gen BRCA2 confieren un 7.3-8.6 veces más riesgo a desarrollar cáncer de próstata antes de los 65 años, comparado con los no portadores de mutación 28. En pacientes jóvenes la prevalencia de variantes germinales en el gen BRCA2 es de 2.9% confiriendole 23 veces más riesgo a desarrollar cáncer de próstata hasta los 56 años 28.
En los casos asociados al síndrome de Lynch, debido a variantes en los genes MMR involucrados en la reparación de los errorses de la replicación del ADN, el riesgo de cáncer de próstata es 3 veces mayor comparado con la población en general 27.. Además se han reportado variantes germinales en otros genes reparadores del ADN como ATM, CHEK2, PALB2, RAD51D, RAD51C entre otros 30..
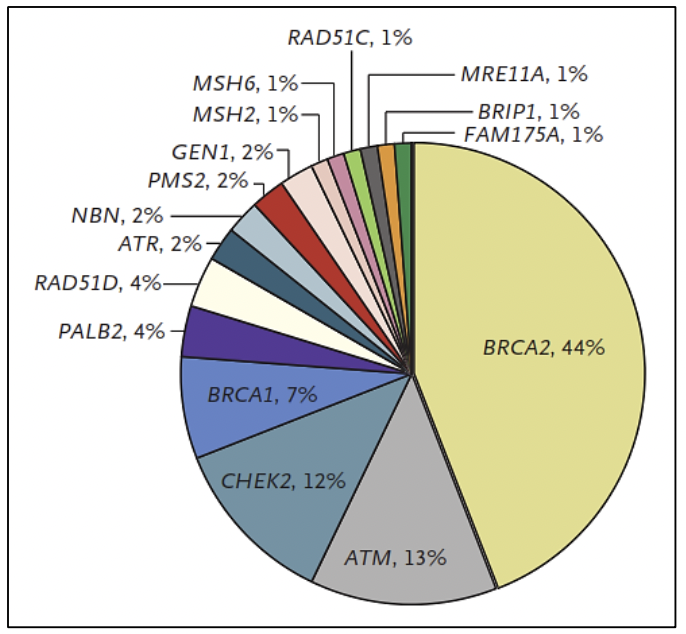
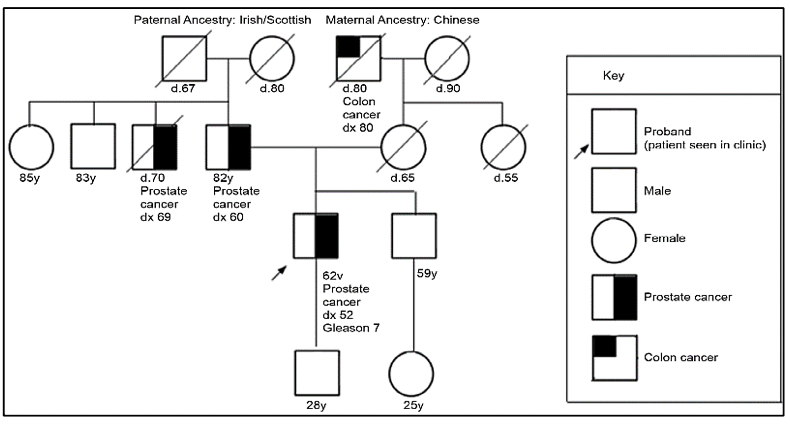
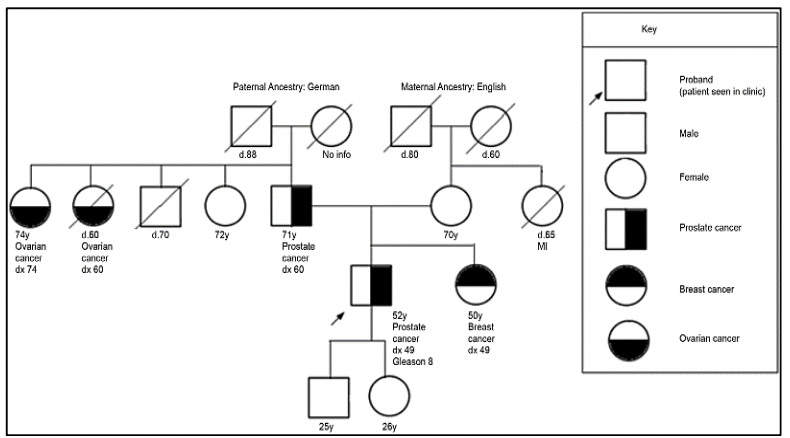
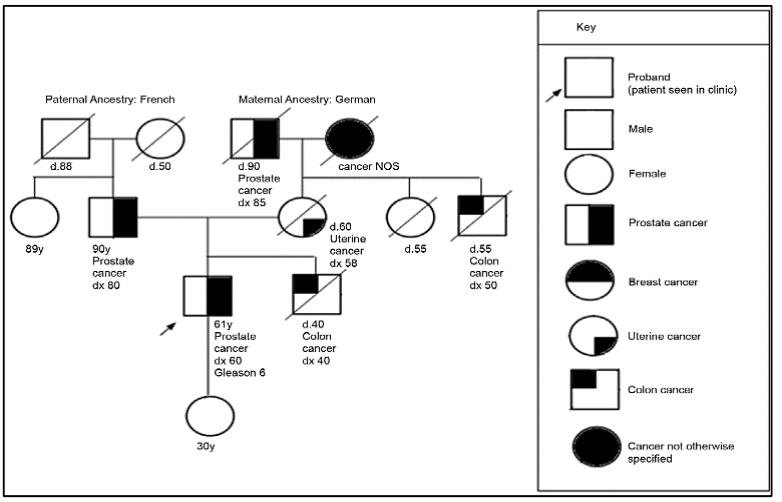
CONCLUSIÓN
Conocer las alteraciones moleculares del cáncer de próstata, así como su clasificación molecular, heterogeneidad genómica y su evolución clonal, permite un manejo adecuado de los pacientes. Además, la recopilación de los datos de la historia familiar del paciente permite sospechar en una predisposición genética al cáncer de próstata y así referirlo para una asesoría genética oportuna y poder brindarle las opciones de seguimiento y terapéuticas.
Contribuciones de Autoría: La autora participó en la génesis de la idea, diseño de
proyecto, recolección e interpretación de datos y preparación del manuscrito del presente
trabajo de investigación.
Financiamiento: Autofinanciado.
Conflictos de intereses: La autora declaran no tener conflictos de interés en la
publicación de este artículo.
Recibido: 15 de Mayo 2022
Aprobado: 06 de Julio 2022
Correspondencia: Maria del Carmen Castro Mujica.
Dirección: Av. Alfredo Benavides 5440, Santiago de Surco, Lima-Perú.
Teléfono: 987597107
Email: mc.castro.mujica@gmail.com
Artículo publicado por la Revista de la Facultad de Medicina Humana de la Universidad Ricardo Palma. Es un articulo de acceso abierto, distribuido bajo los términos de la Licencia Creatvie Commons: Creative Commons Attribution 4.0 International, CC BY 4.0(https://creativecommons.org/licenses/by/1.0/), que permite el uso no comercial, distribucion y reproducción en cualquier medio, siempre que la obra original sea debidamente citada. Para uso comercial, por favor póngase en contacto con revista.medicina@urp.edu.pe.
REFERENCIAS
