INTRODUCTION
Amyloidosis, formerly called primary systemic amyloidosis, is a disease caused by the extracellular deposit of insoluble proteins with a fibrillar structure composed of subunits of low molecular weight, most of which is between 5 to 25 kDa, being conditioned by alterations in the folding of these proteins. It has a positive staining for Congo red and apple green birefringence in polarized light microscopy. Commonly, it predominates in males and although the physical findings are not characteristic, frequent symptoms are fatigue and weight loss. However, macroglossia is one of the most specific signs. In addition, the syndromic presentation frequently involves the kidney with proteinuria in the nephrotic range with or without renal and heart failure. When cardiac problem is present, congestive heart failure is due to restrictive cardiomyopathy. Sometimes it associates gastrointestinal and nervous system problems1,2,3,4.
Among the types of amyloidosis, the primary and secondary types are described. In primary amyloidosis (AL) the protein deposit is derived from fragments of immunoglobulin light chains (the variable portion) and is found in the group of plasma cell dyscrasias with clonal expansion. The secondary form (AA) occurs as a complication of chronic diseases such as rheumatoid arthritis (RA), spondyloarthropathies, osteomyelitis and tuberculosis1.
CASE REPORT
A 47-year-old male patient admitted to the nephrology department at Almenara Hospital with an 18-month disease, characterized by foamy urine, progressive weight loss and edema of the lower limbs. He is diagnosed with chronic kidney disease (CKD) receiving medical treatment. Four months before admission, I started hemodialysis therapy. For the past 2 months, he had bilateral hand stiffness, which starts on the first finger of both hands, progressively extending to the third finger with limited functional activity and does not report dysesthesias.
It has a weight loss of approximately 10Kg in the last two weeks. On admission, she reported dysphagia only to liquids, dysarthria, pain in the oral cavity and swallowing; persistence of foamy urine. He denies edema, vomiting, early fullness, diarrhea or fever. No medical history of importance, but his father had diabetes and CKD, maternal aunt and cousin with stage 5 CKD in hemodialysis therapy (3 times a week) did not need the cause.
Physical examination at admission: blood pressure 120/80 mmHg, heart rate 80 beats/min., respiratory rate 18/min., weight 53 Kg, pallor, no edema, thin skin with muscular atrophy. Oral cavity with macroglossia. No pulmonary, cardiovascular compromise or presence of visceromegaly. In the locomotor examination the tinnel sign was negative and, in both hands, he had bilateral stiffness with limited mobility
Lab test results showed CBC with leukocytes 9210/mm3, lymphocytes 2210/mm3, bands 276/mm3, monocytes 276/mm3, Hb: 6.67 g/dl, VCM: 97 fL, HCM: 32 pg, viscosidad sérica 4´5”, creatinine: 7.16 mg/dl, urea: 56 mg/dl, glucose 69 mg/dl, phosphatase alkaline: 67 U/L, TGP: 13 U/L, albumin: 3.42 gr/dl, TGP: 13 mm/H, platelets: 353 mil/mm3, fibrinogen: 3.7 g/L, PTTa: 27” PT: 9.7” INR: 0.93. Complete urine test: Density 1000, pH 8, glucose +++, thevenon +++, leukocytes 0-14 xc, RBC 1-2 xc. Uric acid 6.68 mg/dL, phosphorus 1.97 mmol/L, total calcium 3.58 mmol/L, ionic calcium 1.47 mmol/L, RF 4 IU/mL, PCR 12.5 mg/L, ferritin >1500 ng/mL, Ca19-9 2.93 U/mL, Ca15-3 8.06, AFP 3.37 ng/mL, CEA 0.99 ng/mL. β2-microglobulin 31.6 mg/L, proteinuria 3186 mg/24h, markers for hepatitis B and C were negative. Electrophoretic plasma proteinogram with moderate hypoproteinemia and increase of alpha1, alpha2 and beta and gamma decrease. Electrophoretic proteinogram in urine with marked non-selective proteinuria with a positive immunofixation for Bence Jones.
Ultrasound showed both kidneys findings compatible with CKD. In the abdominopelvic TEM, no tumors or adenopathies were reported. Upper gastrointestinal endoscopy shows distal erythematosus esophagitis with indirect signs of gastroparesis. The aspiration biopsy of subcutaneous fatty tissue with Congo red was negative. During its evolution the patient course with an in-hospital pneumonia and dies.
The pathological diagnosis was a systemic amyloidosis with mediastinal, pericardial, pulmonary and renal involvement as shown in Figure 1.
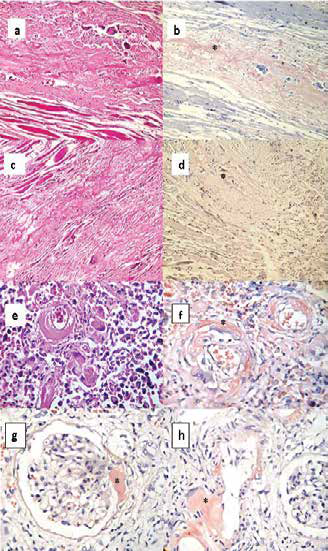 Figure 1.
Figure 1. Histopathology in hematoxylin-eosin and Congo red (+) (in asterisk) in pre-laryngeal muscle (a and b), pericardium (c and d), lung (y and f ) and kidney (g and h). The deposit of acellular amorphous hyaline material and the presence of giant multinucleated cells foreign-body like in the muscle fibers (a), in the pericardium with areas of fibrosis (c), in the lungs at the perivascular level (e), and in kidneys in mesangio, vascular pole and blood vessels (g). Congo red (+) (b, d, f, g and h).
DISCUSSION
The present case was constituted by an adult patient without a personal history of chronic diseases, but a positive family history of chronic kidney disease (maternal line) in which some form of hereditary familial amyloidosis could be thought, whose dominant heterozygous disorder replaces a protein amino acid making it amyloidogenic with transthyretin being the most common amyloidogenic protein. The prognosis of life is longer, unlike this case, which had a survival of 4 months and there was macroglossia, which is not usually described in the familiar form2,3,6.
The absence of chronic inflammatory diseases did not support a form of secondary amyloidosis (AA). The patient had started dialysis therapy 4 months ago, which meant that there was no possibility of amyloidosis associated with dialysis. This is generally described in patients with dialysis treatment greater than 10 years and is the result of deposition of fibrillar structures derived from β2 microglobulin with the presence of typical signs such as the painful shoulder due to amyloid infiltration in the synovial membrane in the glenohumeral joint5. The clinical form of this case, rather, was manifested with chronic renal disease evolving to stage (5) associated with proteinuria in more than 3g, without severe hypoalbuminemia or edema, and residual diuresis. In patients with amyloidosis, the kidney is the most frequently affected organ. Usually 75% of patients with amyloidosis have proteinuria and 30% of these in nephrotic range. However, only 30% of patients with renal amyloidosis will require dialysis. The average time from diagnosis to the dialysis onset is 14 months and the survival time after starting dialysis therapy is 8 months1,2,6. In this case, there was just a history of clinical manifestations of nephrotic syndrome 14 months before starting dialysis therapy and his survival after starting dialysis was 4 months.
It has been suggested that there is an increase in renal size in amyloidosis, as in effect occurs due to amyloid deposit, nevertheless, it is reported that in the majority of patients the kidney size is norma2,4, and in this case they were described hypotrophic kidneys associated with CKD. In relation to plasma cell dyscrasia, in monoclonal gammopathies such as multiple myeloma, there is a usual relationship of kappa light chains: lambda, which is reversed in amyloidosis with a preponderance of the lambda chain in 3.5: 1.
About the extrarenal manifestations of amyloidosis, the second most frequently affected organ is the heart which is associated with a myocardiopathy with diastolic dysfunction. It runs with a pseudo-infarct pattern with QS complexes. Echocardiography is important as a diagnostic aid and may describe thickening of the ventricular myocardium wall without a history of hypertension which should lead to consideration of an infiltrative process such as amyloidosis. This finding does not affect survival, unless it is associated with heart failure or septal thickening in more than 15 mm. This patient did not undergo an echocardiography and had no clinical history of heart failure; however, it should be considered that the main cause of death of patients undergoing renal replacement therapy are heart problems. Pericardial infiltration was described under microscopy, but it should be emphasized that this rarely results in constrictive pericarditis1,2,6.
The patient did not course with liver involvement (where an association with the increase in alkaline phosphatase is described), there was no hepatosplenomegaly (hepatomegaly is described in 25% of patients and splenomegaly is unusual). There was a consumptive syndrome probably associated with a malabsorption syndrome. It is worth mentioning that in patients with gastrointestinal compromise, most of them are asymptomatic, with diarrhea-associated malabsorption in less than 5% so not all of them have diarrhea. Although it is described that there is usually the presence of vascular deposits in the body, the submucosa (which was not described in this patient), the mechanisms associated with malabsorption are not fully established since they may include processes such as autonomic neuropathy of the myoenteric system, myopathy secondary to smooth muscle infiltration or ischemia secondary to vascular infiltration1,2,6,7. It is important to note that in this patient indirect signs of gastroparesis, probably associated with neuropathic compromise, were described at the endoscopic level.
Regarding the involvement of the nervous system associated with amyloidosis, carpal tunnel syndrome occurs in about half of patients and symmetric sensory distal to proximal neuropathy is often described, sometimes associated with dysesthesias. The involvement of the median nerve by the symptomatic distribution of it was suggestive in this patient. At the hematological level, it had a normal coagulation profile and there was no thrombocytosis (which is described secondary to hyposplenism in relation to amyloid infiltration). The compromise at the level of coagulation is usually described by deficiency of factor X in relation to its binding with amyloid fibers, reduction of coagulation factors due to advanced hepatic involvement or by altered fibrinolysis due to deficiency of alpha 2 antiplasmin1,2,4,6. The respiratory compromise in these patients is usually asymptomatic, although in this case there was an amyloid infiltration, as well as at the level of the prelaryngeal muscles.
Finally, in relation to clinical presentation, it is important to differentiate that in the case of AA amyloidosis, the cardiac component is unusual and may present with hepatosplenomegaly. In the rest of the diseases with immunoglobulin deposit not amyloid (heavy chain deposit disease, etc.), the progression is slower, and macroglossia does not occur1,2,4.
The diagnosis of this patient was correlated with non-selective proteinuria with immunofixation for the Bence Jones lambda protein. In patients with amyloidosis, the initial screening of the M protein (monoclonal) must be done with immunoelectrophoresis and immunofixation in serum and urine, which will be detectable in 90% of cases. It is reported that serum immunofixation has a sensitivity of 69% versus urinary immunofixation that has a sensitivity of 83% and that was positive in this patient. If monoclonal proteins are not found by immunofixation technique, the quantification of free light chains is suggested, which has a sensitivity of 91% in cases of AL amyloidosis. In patients with senile, familiar or localized amyloidosis, no monoclonal proteins are documented4,6,8.
The aspiration biopsy of subcutaneous fatty tissue with Congo red has a sensitivity of 57 to 85% and a specificity of 92 to 100% for both the AA and AL forms. The rectal biopsy has a sensitivity of 84%. Bone biopsy is positive in 30% of patients and gingival biopsy in 50 to 70% of cases. The sensitivity in general will be greater in multi-organ compromise4,6,8. The subcutaneous fatty tissue biopsy of this patient was negative despite multiorgan involvement, but it may be that the marked consumption probably did not allow enough sample of fatty tissue. Finally, at a microscopic level, it is important to know that amyloid AA and LA can be differentiated by specific antibodies. Some time ago, pretreatment with potassium permanganate was done, which eliminated Congo red positive for AA amyloid, but not for AL amyloid9.
Thus, for the diagnosis of LA, the following 4 criteria will be required: 1. Presence of a systemic syndrome related to amyloid. 2. Congo red positive stain on samples such as fatty aspirate, bone marrow or organ biopsy. 3. Direct examination (immunoperoxidase staining, sequencing) that reveals the amyloid related to light chains. 4. Evidence of cellular monoclonal expansion (as serum or urinary monoclonal protein, plasma cells in bone marrow)6.
The use of proteosome inhibitors, immunomodulatory drugs and monoclonal antibodies, and autologous stem cell transplantation has been described, which has improved the prognosis of patients with a low to intermediate commitment, however, for those with greater commitment the prognosis is still bad. The early diagnosis will be determinant for the effectiveness of the treatment and prognosis, due to the natural progression of the diseasesup>3.
Authorship Contributions:The author participated in the conception and design of the work; data collection / collection; statistical contribution; data analysis and interpretation; critical review of the manuscript; writing of the manuscript and approval of its final version.
Financing: Self-financed.
Interest conflict: The author declares no conflict of interest in the publication of this article.
Received: December 02, 2018.
Approved: January 22, 2019.
Correspondence: Edwin Rolando Castillo Velarde
Address: Av. Miguel Grau 800, La Victoria 15033. Lima-Peru.
Telephone: +513242983
E-mail: javiercdc@gmail.com
BIBLIOGRAPHIC REFERENCES
2.- Gertz, Morie A. MD Amyloidosis. Monoclonal gammopathies and related disorders. Hematology, Oncology Clinics of North America. 1999; 13, Issue 6: 1211-33
3.- Vaxman I Gertz M, Recent Advances in the Diagnosis, Risk Stratification, and Management of Systemic Light-Chain Amyloidosis, Acta Haematol 2019; 141: 93–106.
4.- Gertz M, CME Information: Immunoglobulin Light Chain Amyloidosis: 2016 Update on Diagnosis, Prognosis, and Treatment, American Journal of Hematology 2016; 91 (9): 948-56.
5.- William L. Henrich. Diálisis. Amiloidosis por microglobulina B2 en la Nefropatía Terminal. 2º edición. México: McGraw Hill; 2001, p. 345-55.
6.- Leung, Nelson MD. Renal Manifestations of Plasma cell Disorders. American Journal of Kidney Diseases. 2007; 50 (1): 155-65.
7.- Suzanne R. Hayman, MD. Primary Systemic Amyloidosis: A cause of Malabsorption Syndrome. Am J Med. 2001; 111:535-40.
8.- Jacques Wallach. M.D. Interpretación Clínica de las pruebas de laboratorio. Amiloidosis. 4º edición. Barcelona: Masson; 2002. p. 1160-63.
9.- Sternberg's. Diagnostical Surgical Pathology. 4º edition, Phildadelphia: Lippincott Williams & Wilkins; 2004. p. 1905-09

http://bit.ly/2Jv5GAc
¿ Quieres dejar tu comentario o sugerencia sobre este articulo?
--> CLICK AQUI <---



