
3rd LATINOMARICAN CONFERENCE IN LIFESTYLE MEDICINE
INICIB

DOCENTES INVESTIGADORES EN REGINA

Latin American Lifestyle Medicine Association
CASO CLÍNICO
DOI 10.25176/RFMH.v19i3.2153
1Instituto Nacional de Salud del Niño San Borja, Lima-Perú.
2Hospital Nacional Edgardo Rebagliati Martins, Lima-Perú.
3Instituto de Investigación en Ciencias Biomédicas (INICIB), Universidad Ricardo Palma, Lima-Perú.
aMédico asistente.
bMédico asistente unidad de cardiología pediátrica.
cMédico ocupacional.
dMédico Investigador.
RESUMEN
El presente caso corresponde a un paciente de 1 mes de vida, sexo masculino con diagnóstico de miocardiopatía no compactada asociada a defectos cardiacos congénitos.
La miocardiopatía no compactada recién es incluida por la AHA como entidad propia a partir de la segunda mitad de la década pasada. El diagnóstico principalmente es ecocardiográfico. La sintomatología en menores de un año puede empezar con falla cardiaca. La evolución es variable con tendencia a la mejoría en algunos casos para finalmente en décadas posteriores se hace más pronunciada la falla cardiaca, eventos tromboembólicos, arritmias malignas y muerte súbita. El manejo esta en medicamentos para falla cardiaca, evitar arritmias malignas y eventos tromboembólicos.
Palabras clave:Miocardiopatía no compactada, defectos cardiacos congénitos, falla cardiaca, ecocardiografía.
ABSTRACT
The present case corresponds to a 1-month-old male patient with a diagnosis of non-compacted cardiomyopathy associated with congenital heart defects. Noncompacted cardiomyopathy is newly included by the AHA as its own since the second half of the last decade. The diagnosis is mainly echocardiographic. Symptoms in children under one year may start with heart failure. The evolution is variable and tends to improve in some cases. Finally, in later decades, heart failure, thromboembolic events, malignant arrhythmias and sudden death become more pronounced. The management is in medicines for heart failure, to avoid malignant arrhythmias and thromboembolic events.
Keywords: Non-compacted cardiomyopathy, congenital heart defects, cardiac failure, echocardiography.
INTRODUCCIÓN
La miocardiopatía no compactada representa una detención en el proceso de compactación miocárdica, caracterizándose por la presencia de múltiples trabeculaciones y de recesos profundos entre trabeculaciones, además de una capa compactada delgada1.
Dicho proceso de compactación ocurre normalmente entre la 5 – 8 va semana de vida fetal y se caracteriza por la compactación
gradual del miocardio, transformación de grandes espacios intertrabeculares en capilares y
evolución de la circulación coronaria. Dicho
proceso típicamente progresa de epicardio a endocardio y de base a ápex2.
La verdadera incidencia de miocardiopatía no compactada es desconocida como resultado de los criterios diagnósticos y nomenclatura inconsistente3.La prevalencia de miocardiopatía no compactada es 0.01% en adultos y 0.14% en niños4.
La miocardiopatía no compactada se ha identificado como la más frecuente luego de la miocardiopatía dilatada e hipertrófica, representando entre 5-9% de las cardiomiopatías en niños5.
La miocardiopatía no compactada se puede presentar de forma aislada, asociada a cardiopatía congénita, con componente genético y hereditario entre 18 – 50%5.
La ecocardiografía se considera de referencia para el diagnóstico. Chin et al en los años 90 dio las pautas iniciales6 de una patología poco entendida e incluida en la clasificación de miocardiopatías por la AHA a partir de la segunda mitad de la década pasada7.
El conocimiento de esta patología es importante para planificar el manejo, pronóstico y valorar el estudio familiar, además de valorar patología extracardiaca.
REPORTE DE CASO
Lactante menor previamente sano que 1 día antes del ingreso de manera brusca presenta pobre lactancia, irritabilidad y dificultad respiratoria, ingresa por emergencia Hospital cercano, encontrando frecuencia cardiaca de 280, recibe amiodarona y remite el cuadro. El paciente es trasladado al HNERM para estudio. donde presenta 2 episodios de TSVP, remitiendo con adenosina, quedando hospitalizado en Unidad de Cuidados Especiales hasta su estabilización hemodinámica. Antecedentes personales: Peso al nacer 3308 g, edad gestacional 38 semanas, lactancia mixta, ganancia ponderal de 29.7 g/d. Antecedentes familiares: Hermanos no antecedentes de cardiopatía. Padre de 38 años sano, madre de 43 años sana. Examen físico: Peso: 4.2 kg, FC: 146, FR: 58, T: 36.6 ºC, PA: 85/65 mmHg, STO2: 98% (FIO2 21%). No dismorfismo, despierto, ventilando espontáneamente, llenado capilar menor de 3 segundos. Ap resp: No rales. Ap cv: Precordio normodinámico, RCRR, SS II/VI BEI, pulsos periféricos palpables, de buena intensidad. Abdomen: Hígado 2.5 cm debajo del reborde costal derecho. Neurológico: Fontanela normotensa, tono y reflejos adecuados.
Pruebas complementarias
Figura 1Radiografía tórax: ICT: 0.62, flujo pulmonar aumentado
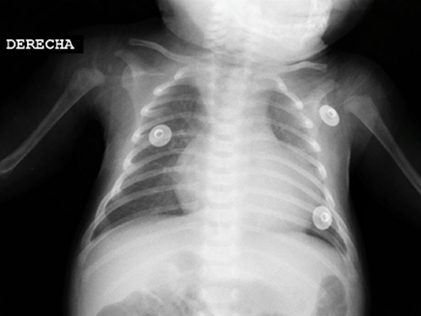
Figura 2. EKG: Ritmo sinusal, FC: 150, PR: 120 ms, QRS: 60 ms, QTc: 440 ms, < QRS 0⸰. signos de crecimiento ventricular izquierdo.

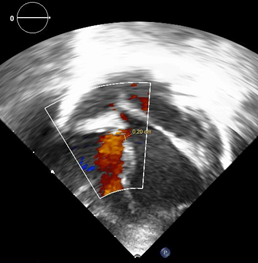
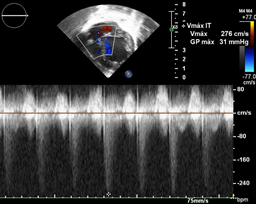
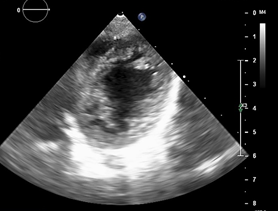
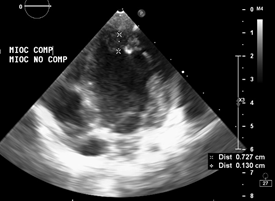
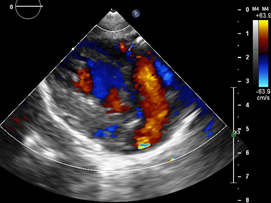
Ecocardiografía: DDVD: 16, DDSIV: 3, DDVI: 28, DSVI: 19 mm (+3DE), DPPVI: 3, R Ao: 11 mm, AI: 13 mm.(0.05 DE), FE: 42% ( Simpson), gradiente de regurgitación tricuspídea 31mmHg (figura 5), gradiente V Ao 0.9 m/s (3 mmHg), gradiente VP 1.3 m/s (8mmHg), presión sistólica de arteria pulmonar 36 mmHg anillo mitral: 19.3 mm (+2DE), anillo tricuspídeo: 20.7 mm (+2DE). Situs solitus, Levocardia, 4 venas drenan en AI, suprahepáticas colapsan bien, VCI y VCS drenan AI. Concordancia AV, regurgitación tricuspídea y mitral leve. Concordancia VA, válvulas sigmoideas competentes .Ventrículo izquierdo dilatado, con SIV desplazado a la derecha, más de 3 trabeculaciones a nivel de la punta del VI y pared ínferolateral y anterolateral (figuras6,7,8), con recesos entre las trabeculaciones y entrada de flujo sanguíneo, miocardio no compactado/ compactado de 5.5 (figura9,10). Coronarias normales. Comunicación interventricular de 2 mm shunt de izquierda a derecha (figura3), Comunicación interauricular de 2 mm shunt izquierda a derecha (figura 4), PCA de 1.5 mm. Laboratorio: Glucosa: 97 mg/dl, urea: 15 mg/dl, creatinina: 0.26 mg/dl, Hb: 12 mg/dl, plaquetas: 346 000, TP: 15, TTP: 49, troponina I: 0.373, CreatininKinasa 192 U/l, CK -MB: 5.47 ng/ml, VSG: 19 mm/h fibrinógeno: 238 mg/dl calcio: 10 mg/dl, fósforo: 5.4 mg/dl, magnesio:2 mEq/l, sodio: 138 mEq/l, K: 5 mEq/l, cloro: 109 mEq/l.



Diagnóstico definitivo:
Miocardiopatía no compactada juvenil, asociada a defectos cardiacos congénitos.
Diagnóstico diferencial
Miocarditis
Miocardiopatía dilatada
Miocardiopatía secundaria a arritmia cardiaca
Manejo
Se inició manejo de falla cardiaca con diuréticos, inhibidor ECA, betabloqueador y agente antiagregante plaquetario.
Discusión:
La miocardiopatía no compactada viene siendo reconocida como entidad separada de las otras miocardiopatías7. A la fecha se cree que es debido a una detención en la embriogénesis normal del corazón, con detención del proceso de compactación8. Conocer la etiología de la enfermedad se encuentra limitada por la heterogeneidad genética, con un limitado entendimiento de la regulación de la trabeculación miocárdica y compactación8.
A la fecha no solo se considera que sería causada por alteración genética, se ha encontrado transmisión familiar en 18 -50% de pacientes adultos, con variabilidad fenotípica a lo largo del tiempo, pudiendo cambiar a cardiomiopatía dilatada o hipertrófica9. También se ha encontrado el desarrollo de hipertrabeculación en pacientes jóvenes deportistas10 y en mujeres gestantes11 con reversión posterior, considerando que esta enfermedad podría ser también adquirida.
Estudios genéticos han demostrado que la deficiencia de la proteína citoplasmática FKBP 12 asociada a los receptores BMP/ activina/ TGFB1 conducen a una deficiente regulación de la trabeculación y compactación del miocardio ventricular8 y conlleva entonces a una hipertrabeculación. A la fecha los genes involucrados en la no compactación son FbKp1a, G4.5/ proteína TAZ, delección cromosoma 14-3-3, proteína ZASP, proteína TNNT2, proteína MYH7, proteína TPM1, proteína MYBPC3, proteína ACTC1, entre otros12.
El caso presentado cumple los criterios ecocardiográficos propuestos por Jenni et all1, Chin el all6 y Stöllberger et all13 y más recientemente lo propuesto por Lai et all3: Apariencia de dos capas de miocardio con capa compacta delgada y capa no compacta gruesa, incremento del número de trabeculaciones pared lateral y ápex VI, trabeculaciones que se mueven sincrónicamente con el miocardio, perfusión de los recesos intertrabeculares y función ventricular anormal.
La edad de presentación es variable, pero con mayor cantidad de diagnósticos en menores de un año y predominantemente de sexo masculino14 como en el caso presentado.
La presentación clínica va desde asintomático15, falla cardiaca, arritmias16, eventos tromboembólicos y muerte súbita. Nuestro paciente se presentó con falla cardiaca más taquicardia supraventricular.
La radiografía de tórax muestra cardiomegalia e hiperflujo pulmonar, no específica de esta patología. El estudio EKG muestra signos de crecimiento ventricular izquierdo, prolongación del segmento QT, datos que también son reportados en la literatura16. La herramienta diagnóstica clave es la ecocardiografía, sin embargo, se debe considerar que ningún parámetro es Gold Estándar ya que sólo valoran parámetros morfológicos. Se consideran las mejores vistas paraesternal eje corto y apical 4 cámaras, pero no se debe dejar de lado las otras vistas. El segmento más comprometido es el ápex ventricular, los otros segmentos varían de acuerdo al estudio17; sin embargo, hay que considerar que mientras más segmentos estén comprometidos existe un peor pronóstico18,19.
La valoración funcional del miocardio no compactado en pacientes asintomáticos se podría hacer valorando el strain longitudinal por la técnica de speckle tracking20,21 y así determinar precozmente disfunción ventricular y tratamiento precoz.
El manejo del paciente con miocardiopatía no compactada asilada o asociada a defectos cardiacos congénitos, como en nuestro paciente, se encamina a manejar la falla cardiaca extrapolado de la miocardiopatía dilatada ya que no existen estudios prospectivos al respecto12, evitar eventos embólicos sistémicos22 y prevención de arritmias malignas. En el paciente reportado se inició manejo de falla cardiaca y uso de antiagregantes plaquetarios.
El pronóstico de estos pacientes es variable, y se ha reportado que dilatación ventricular, función ventricular disminuida, edad menor de un año, dismorfismos, entre otros constituyen pacientes con peor pronóstico23; aunque en otro estudio menciona como factores de peor pronóstico tener fracción de eyección menor de 50 e hipoplasia de la pared posterior del ventrículo izquierdo24. Según las curvas de seguimiento los pacientes pediátricos que inician con falla cardiaca tienden a mejorar en el tiempo, pero al final se manifiesta un deterioro de la función cardiaca progresiva al igual que el adulto, requiriendo incluso trasplante cardiaco25.
La miocardiopatía no compactada es predomiantemente genética con presentación clínica variable, en algunos casos como el paciente asociado a defectos cardiacos congénitos, por lo que requiere consejo genético, diagnóstico genético y screening cardiológico familiar26.
CONCLUSIONES
-La miocardiopatía no compactada es una entidad nueva de miocardiopatía cuyo diagnóstico va en aumento en los últimos años.
-La miocardiopatía no compactada puede ser aislada o asociada a defectos cardiacos congénitos.
-El diagnóstico de miocardiopatía no compactada es principalmente por criterios ecocardiográficos.
-De presentarse sintomatología en niño pequeño, esta será como falla cardiaca como característica principal.
-La expresión fenotípica de la miocardiopatía no compactada en el tiempo puede cambiar a miocardiopatía dilatada o hipertrófica como variabilidad fenotípica de los genes involucrados.
-El gran porcentaje de carga genética familiar es autosómica dominante y predomina en el sexo masculino.
-El manejo de la miocardiopatía no compactada es extrapolado de la miocardiopatía dilatada y es principalmente manejo de falla cardiaca, prevención de arritmias malignas y prevención de eventos embólicos sistémicos.
-Mientras menor edad al diagnóstico, menor función ventricular y menor pared compacta del ventrículo izquierdo peor pronóstico, requiriendo en algunos pacientes trasplante cardiaco.
Los resultados laboratoriales indican pancitopenia severa leucopenia 2730 K/ul, anemia (Hb: 9.4 g/dl) y plaquetopenia de 66,000 K/ul. Adicionalmente, cursó con electrolitos sodio (Na) 130 mmol/L, CPK (Creatinfosfokinasa) 4135 U/L, CPK-MB 266.42 ng/ml, ProBNP 276 pg/dl y Troponina I: 0.097 pg/dl, en la tomografía de tórax y abdomen (Figura 3), se evidenció derrame pleural bilateral leve, engrosamiento intersticial septal interlobulillar y área de vidrio deslustrado en lóbulos superiores y medios, marco colónico con gran dilatación y neumatosis a nivel de sus paredes, escaso liquido libre perihepático y periesplénico, no evidencia de aire libre.
Dichos resultados en términos generales no son orientativos de alguna patología en particular; los resultados de troponina y el electrocardiograma no son compatibles con el de infarto cardiaco y el de ProBNP tampoco se relaciona con el de Insuficiencia cardiaca descompensada; por el contrario, cada uno de estos resultados se encuentra presentes en el Coma mixedematoso.
Se obtuvo resultados de hormona tiroidea al 3 día: TSH 26.2 Uul/ml T4 libre < 0.30 ng/dl (Tabla 1), confirmando el diagnóstico de coma mixedematoso, posteriormente a los 5 días de su ingreso no respondió adecuadamente al soporte hemodinámico y ventilatorio, falleciendo al cuarto día de internamiento.
DISCUSIÓN
El CM es una entidad clínica de presentación infrecuente, se calcula que la incidencia por año es de 0,22/1000,000 personas al año7; debido al cual puede convertirse en un problema diagnóstico, ya que su poca frecuencia hace que el evaluador inicial puede no plantearlo como un diagnóstico desde el inicio.
En el pasado la tasa de mortalidad global por mixedema era del 60 al 70% de casos; el reconocimiento temprano y los avances en el soporte de atención intensiva han reducido la tasa de mortalidad a 20 – 25%8; que aún es alto.
Cheng y colegas, en un estudio notaron que hacer un diagnóstico temprano de un evidente hipotiroidismo primario en el servicio de urgencias es difícil: solo el 21% de los pacientes ingresaron con una impresión diagnóstica inicial correcta9. Quizá lo más preocupante fue que el 50% de los pacientes del estudio que finalmente fueron diagnosticados con mixedema no fueron diagnosticados en el departamento de emergencia. Este estudio indica que la crisis hipotiroidea no es bien reconocida por el médico de emergencia. En el caso de nuestro paciente se tardó 24 horas para plantearse la posibilidad diagnóstica de Coma mixedematoso.
Los pacientes con coma mixedematoso generalmente se presentan en los meses de invierno, lo que sugiere que el frio externo puede ser un factor agravante10; coincide con el caso de nuestro paciente que se presentó en el mes de julio, que es la estación de invierno y uno de los meses más fríos de esta ciudad.
La epidemiología de la crisis mixedematosa sigue el mismo patrón como en el hipotiroidismo y es más común en mujeres y ancianos11. El hipotiroidismo es cerca de 10 veces más frecuente en mujeres que en hombres 12. En el reporte de caso el paciente fue de sexo masculino de 82 años, hecho que pudo haber contribuido al retraso del planteamiento como diagnóstico probable.
El paciente hipotiroideo se presenta mayormente en el departamento de emergencia con múltiples quejas vagas de inicio insidioso, y tienen un examen físico inespecífico, circunstancias que pueden hacer el diagnóstico esquivo aun para el medico acucioso. Los signos y síntomas presentes a menudo varían con la edad y género y la severidad de estas características clínicas varían grandemente13. Caso típico del paciente en discusión que tuvo una evolución clínica completamente inespecífica ya que la escasa información obtenida a través de una anamnesis indirecta no permitió contar con los antecedentes del paciente y la falta de datos en su registro no contribuyó a un adecuado enfoque. El hipotiroidismo debe ser siempre considerado en pacientes quienes se presentan con síntomas no específicos de enfermedad sugestiva, incluyendo debilidad, intolerancia al frío, y alteraciones en el estado mental, y de recibir drogas que deterioran la función tiroidea o tratamiento de cáncer avanzado de cabeza y/o cuello. Adicionalmente, pacientes con falla cardiaca crónica estable o efusión pericárdica inexplicable justifican pruebas tiroideas séricas9.
Las infecciones y septicemia son los principales factores precipitantes8. Las infecciones típicamente incluyen neumonía, infecciones del tracto urinario y celulitis. Accidentes cerebro vascular, falla cardiaca congestiva, accidentes de tráfico, sangrado gastrointestinal, y varios grupos de sedantes pueden jugar un rol importante en la precipitación de crisis mixedematosa11. Un factor de fondo comúnmente ignorado en la crisis mixedematosa es la descontinuación de suplemento tiroideo en pacientes críticamente enfermos. En el caso presentado, por lo menos el paciente no tenía antecedente de padecer patología tiroidea.
El diagnóstico de Coma mixedematoso es principalmente clínico. La presencia de estupor marcado, confusión o coma, e hipotermia en un paciente con hallazgos de hipotiroidismo es fuertemente sugestivo de coma mixedematoso. El examen físico es demostrativo de hipotiroidismo: piel seca, gruesa y escamosa, cabello escaso o áspero; edema de piel y partes blandas, macroglosia, voz ronca y reflejos tendinosos profundos retardados. Otras características clínicas importantes del coma mixedematoso incluyen hipoventilación, bradicardia, disminución de la contractilidad cardiaca, disminución de la motilidad intestinal, íleo paralítico y megacolon12. Lo resaltante del caso presentado, es que sorprendentemente presentaba todas estas características clínicas al realizar el examen físico; piel fría, edema, presión arterial conservada que evolucionó a hipotensión; bradicardia; abdomen distendido (íleo paralítico), un estado neurológico no óptimo: Escala de Coma de Glasgow (ECG) al ingreso de 12 puntos que luego bajo a 7 puntos que hizo necesario la necesidad de intubación endotraqueal y soporte ventilatorio por la hipoxemia e hipercapnia que presentaba.
Reinhardt y Mann reportaron hipoxemia en 80%, hipercapnia en 54%, e hipotermia con una temperatura menor de 34.5°C en el 88% de todos los pacientes con crisis mixedematosa14.
Así mismo, bradicardia sinusal, complejos de bajo voltaje, bloqueo de rama, bloqueo cardiaco completo, y cambios inespecíficos del ST en el electrocardiograma fueron registrados en la crisis mixedematosa10. Nuestro paciente presentaba una bradicardia sinusal con un QT prolongado.
Un importante aspecto práctico puede ser la identificación de efusión pericárdica e infarto del miocardio en un escenario de crisis mixedematosa. Los complejos de bajo voltaje y los cambios los inespecíficos del ST pueden ser vistos en la efusión pericárdica. Deben realizarse enzimas cardiacas ante sospecha de infarto11. Se halló efusión pleural bilateral más no efusión pericárdica, y se tuvo la sospecha de infarto cardiaco por lo que se solicitó troponina la que resultó negativa, en un segundo momento se planteó disfunción del nodo sinusal, que luego de evaluación por cardiología la descartó y lo consideró de origen metabólico.
El TSH elevado, y las concentraciones séricas muy bajas de T4, FT4 y T3 confirman el diagnóstico de Coma mixedematoso. Otros hallazgos laboratoriales incluyen anemia, hiponatremia, hipercolesterolemia, lactato deshidrogenasa sérica elevada, concentraciones de CPK elevadas. Los gases de sangre arterial pueden revelar hipoxemia, hipercapnia y acidosis12. Cada uno de estos resultados mencionados lo presentaba nuestro paciente; en relación al perfil tiroideo es importante destacar que en nuestra emergencia no contamos como prueba de rutina y para realizarlo tenemos que tener la autorización de un endocrinólogo o del patólogo clínico encargado de laboratorio y los resultados se expiden al tercer día de tomadas la muestra.
Los pacientes con Coma mixedematoso deben ser manejados en una Unidad de Cuidados Intensivos, bajo monitoreo continuo, especial atención debe darse al soporte ventilatorio en estos pacientes y la ventilación mecánica debe darse si se requiere. La hipotermia e hipotensión deben ser corregidas. Los disturbios metabólicos tales como hiponatremia, hipoglicemia e hipercalcemia, los cuales pueden agravar el estado mental alterado deben corregirse. Se debe de investigar por todos los factores precipitantes del coma mixedematoso. Asimismo, se deben extraer los cultivos y tomar radiografías de tórax para descartar infecciones, si están presentes, deben tratarse de manera agresiva con un tratamiento antibiótico adecuado12. Nuestro paciente ingresó por la Unidad de Shock Trauma de donde fue derivado al área de UCINE (Unidad de Cuidados Intermedios de Emergencia) donde recibió el soporte ventilatorio invasivo y apoyo de fármacos vaso activos pertinentes; no pudimos corroborar un factor infeccioso precipitante del Coma mixedematoso: urocultivo negativo, tomografía de tórax negativo para neumonía, no evidencia de lesiones dérmicas; se planteó una infección con punto de partida abdominal por la gran dilatación de asas intestinales gruesas con neumatosis y liquido libre perihepático.
El uso empírico de glucocorticoides debe ser parte del protocolo terapéutico inicial, que indican que el hipotiroidismo grave induce una menor respuesta adrenal al estrés, independiente de que haya o no insuficiencia adrenal simultánea. Dado que la hormona tiroidea acelera el metabolismo del cortisol, los glucocorticoides deberán administrarse siempre antes del reemplazo tiroideo, ya que lo contrario podría precipitar una crisis adrenal. Se administrará hidrocortisona en dosis de estrés, 50-100 mg por vía intravenosa (IV) cada 6-8 h, durante 7 a 10 días o hasta estabilizar hemodinámicamente al paciente3. Nuestro paciente recibió hidrocortisona 100 mg c/8 horas.
Deben administrarse dosis altas de levotiroxina (LT4), con el objetivo de reemplazar el déficit y de saturar los depósitos circulantes de hormona tiroidea. La ATA (American Thyroid Association) recomienda iniciar con 200-400 μg en bolo EV en las primeras 48 horas, seguidas de una dosis más fisiológica de 50-100 μg EV diarios hasta poder administrar por vía oral. Si bien algunos proponen comenzar con dosis mayores de 300-500 μg, sería prudente evitarlas en añosos, desnutridos, o con arritmias o infarto de miocardio. Para evitar el riesgo de complicaciones cardíacas realizar monitoreo cardíaco continuo, con reducción de la dosis de hormona tiroidea de observarse cambios isquémicos o arritmias3. Cambiar a la vía oral es posible cuando las condiciones del paciente han mejorado12. En el caso en discusión se indicó levotiroxina 300 ug por sonda nasogástrica por dos días y luego 100 ug por día, (debido a que nuestra institución no cuenta con la presentación de levotiroxina en ampolla y en el Perú no está en el petitorio nacional de medicamentos esenciales del MINSA-2015); su administración se desarrolló seguido de un estricto monitoreo cardiológico, por los riesgos de arritmia o infarto que puede ocasionar dicha administración, aunque nuestro paciente no presentaba en el momento patología isquémica coronaria aguda, ni arritmia.
Ciertamente el Coma mixedematoso trae consigo alteraciones gastrointestinales: como íleo y edema de pared intestinal, al no existir otra alternativa se decidió administrarla por vía digestiva debido a que no se cuenta con presentación en ampollas para la administración endovenosa. Existen revisiones que concluyen que la absorción oral de L-T4 es variable, pero la respuesta clínica ocurre rápidamente aún en el íleo mixedematoso15.
Entre los pacientes tratados con LT4 a dosis de 500 μg/día o más, el resultado de aquellos que recibieron LT4 por vía oral parece más favorable que los tratados con administración intravenosa8,16.
La etiología del hipotiroidismo (primario vs secundario) y la ruta de administración de L-T4 (oral versus intravenoso) no tuvieron influencia sobre los resultados del coma mixedematoso10.
La mayoría de las fuentes, debido a esto, recomiendan el tratamiento EV o levotiroxina oral, ya que la evidencia no parece indicar una diferencia significativa entre los dos13.
Pese al soporte ventilatorio invasivo, la administración de vasoactivos, y la terapéutica en su conjunto; no hubo mejoría del paciente, por el contrario, presentó una evolución tórpida y falleció al cuarto día de hospitalizado.
En el caso presentado lo resaltante de todo es que el paciente presentaba todas las características clínicas y los hallazgos laboratoriales “típicos” de un paciente hipotiroideo descompensado, tal como está mencionado en todas las revisiones bibliográficas; pero lo que hay que destacar es el hecho de que al ser una enfermedad con muy poca frecuencia difícilmente se plantea en un inicio; a menos que el médico evaluador haya visto un caso similar con anterioridad; ello se tornará más dificultoso si el paciente no tiene antecedente de patología tiroidea y si la anamnesis no es la más apropiada, aunque habría que añadir que la sintomatología previa puede resultar muy errática y no orientar a alguna patología específica; otro problema a considerar es el hecho de que las pruebas de laboratorio de perfil tiroideo no se realizan de emergencia rutinariamente y que los resultados suelen emitirse de dos a tres días después. Por otro lado, el no contar con el medicamento en presentación para administración parenteral podría ser perjudicial, aunque algunas publicaciones refieren que no hay mayor diferencia con la administración enteral pese a las alteraciones digestivas que puedan existir en esta patología.
