CLINICAL CASE
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2019 - Universidad Ricardo PalmaDOI 10.25176/RFMH.v20i1.2556
Xp11.2 TRANSLOCATION RENAL CARCINOMA IN A CHILD
CARCINOMA RENAL CON TRANSLOCACIÓN Xp11.2 EN UNA INFANTE
Gloria Paredes Guerra1,a,
Javier Larios León2,b,
Carol Gonzales Gonzales3,b
1 International Clinic. Lima, Peru.
2 Medical Oncology Service, Almanzor Aguinaga Asenjo National Hospital. Chiclayo, Peru.
3 High Complexity Hospital Virgen de La Puerta. Trujillo, Peru.
aPediatric oncologist.
bResident of medical oncology.
ABSTRACT
Translocation renal cell carcinoma (t-RCC) is a subtype of renal cell carcinoma (RCC). This case is about an 11-year-old with pain in right iliac fossa for two weeks. Tumor of 2.2 x 1.9 x 2.8 cm was evidenced in the upper pole of the right kidney, by ultrasound. Then, she underwent to a right radical nephrectomy and regional lymph node dissection, after which CRT subtype Xp 11.2 was diagnosed. Subsequently, first-line treatment with pazopanib and concomitant chemotherapy was initiated.
This disease is more frequent between 10 and 15 years. Their prognosis is usually better compared to adults. The multidisciplinary management associated with the availability of first-line medications improves progression-free survival.
Key words: Carcinoma, Renal Cell; Carcinoma; Translocation, Genetic (Source: MeSH MEDLINE)
RESUMEN
El carcinoma de células renales con translocación (CRT) es un subtipo de carcinoma de células renales (CCR). El presente caso trata de una niña de 11 años con dolor en fosa ilíaca derecha de dos semanas de evolución.
Se evidenció tumoración de 2,2 x 1,9 x 2,8 cm en el polo superior del riñón derecho, por ecografía. Luego, fue sometida a nefrectomía radical derecha y a disección ganglionar regional, tras lo cual se diagnosticó CRT subtipo Xp 11.2. Posteriormente, se inició tratamiento de primera línea con pazopanib y quimioterapia concomitante.
Esta enfermedad es más frecuente entre los 10 y 15 años. Su pronóstico suele ser mejor comparado al de los adultos. El manejo multidisciplinario asociado a la disponibilidad de medicamentos de primera línea, mejora la sobrevida libre de progresión.
Palabras Clave: Carcinoma de Células Renales, Carcinoma, Translocación Genética (Fuente: DeCS BIREME)
INTRODUCTION
Advances in molecular technology have led to better typing of renal cancer types and subtypes. As a result, prognostic and predictive factors are now recognized, leading to an increase in cancer-specific survival.
Renal cell carcinomas are very rare neoplasms in children. They are usually found in adults over age 50. Their incidence in children is estimated to be between 1.8% and 6.3%, and its frequency is directly proportional to age(1).
In 2016, the World Health Organization (WHO) classification of genitourinary tumors added, within the subtypes of renal cell carcinomas, MiT family translocation renal cell carcinoma, whose molecular study encompasses Xp11.2 translocation and translocation t (6; 11), each with variable clinical features and prognoses(1,2).
The management of renal cell carcinomas with translocation is not usually different from other subtypes of renal cell carcinomas. For this type of neoplasia, the primary treatment is surgery, regardless of the finding of metastasis at the onset of the disease. On the other hand, the use of target medications, such as sunitinib or pazopanib, both tyrosine kinase inhibitors (TKI), have contributed to improved overall survival and disease progression-free survival in these patients. Interdisciplinary management and availability of these drugs are essential to ensure long survival in children carrying translocated renal cell carcinoma(1,2).
The case presented in this report highlights the long progression-free survival of a girl who presented Xp 11.2 translocation renal cell carcinoma, determined by fluorescence in situ hybridization (FISH).
CASE PRESENTATION
It is presented the case of a 10-year-old girl, who referred to asthenia, pallor, and “stabbing pain” at the level of the right iliac fossa, not irradiated, of two weeks of evolution, for which she went to the clinic emergency in November 2016.
Her personal history is as follows: post-traumatic renal hematoma of 2.2 x1.9x2.8 cm in April 2013.
2.2 x1.9x2.8 cm in April 2013. On physical examination, it is observed a patient with moderate pallor, no signs of active bleeding, with superficial and deep palpation pain at the iliac fossa and right flank level, no evidence of palpable tumors at the abdominal level. The rest of the examination did not contribute. It is initiated an analgesic treatment with a slight improvement in pain, and it is performed a renal ultrasound, finding, in the upper pole of the right kidney, a solid echogenic image with a hypoechogenic center with regular edges of 33.6 x 22.7 mm, without internal vascularity.
She is also evaluated by the urologist, who requests that a magnetic resonance of the abdomen be attached to the work plan, where is visualized a nodular image of 2.2 x 1.9 x 2.8 cm -which compromises the upper pole of the right kidney-, lobed contours without infiltrative signs, hyposignal in T2 and central hyperintense zone, with slight contrast uptake, compatible with renal tumor.
In the extension study with computerized chest axial tomography, a lymph node conglomerate is evidenced in the path of the azygos vein and the right para-esophageal group. In addition, in the laboratory analyses, 480 ng/dl lactic dehydrogenase, 9.3 g/dL hemoglobin, 9 mg/dL serum calcium, 4200 neutrophils, and 220,000 platelets were found.
Four days later, the patient was stabilized and submitted to a percutaneous renal biopsy, and the anatomopathological result was renal neoplasia of eosinophilic cells.
Because of this diagnosis, it was decided to subject the patient to the right radical nephrectomy associated with regional lymphadenectomy. The anatomopathological findings describe a wine-colored tumor of approximately 8 x 6.5 x 6 cm, with a hard consistency. Under the microscope, a malignant renal neoplasm of predominantly papillary pattern is found, constituted by cells of epithelioid aspect, with high nuclear grade, abundant eosinophilic cytoplasm, involving renal sinus, tumoral necrosis present but minimal, surgical margins free of neoplasm, lymphovascular invasion present, 22 lymphatic nodes resected, of which 7 were infiltrated by malignant neoplasm.
In order to complement this, the following immunohistochemical tests are performed: AMACR (+), CD10 (+), EMA (-), Ki 67%: 15%, CK 7 (-), resulting in translocation renal cell carcinoma.
The genetic study of the tumor is carried out according to the FISH test, and the diagnosis of translocation renal cell carcinoma (t-RCC) was confirmed, with a positive result for translocation Xp11.23/17q25.3 (table 1 and figure 1).
Table 1. FISH Test final result
| Result |
| Translocation Xp11.23/17q25.3: Positive for translocation in 83% of the analyzed cells |
| Translocation Xp11.23/1q23.1: Negative for translocation in 100% of the analyzed cells |
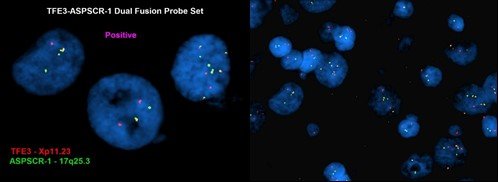
Figure 1. Images from the FISH test that tested positive for the Xp11.2 translocation
Upon confirmation of the diagnosis of translocation renal cell carcinoma, the patient was typed according to the International Metastatic Renal Cell Carcinoma Database Consortium (IMDC) shown in table 2. A score of two points was then obtained (considering a time from diagnosis to treatment less than one year and hemoglobin below the normal lower limit). Finally, it was estimated that the patient belonged to the intermediate risk group.
Table 2. IMDC classification used to typify the patient after radical nephrectomy
| Prognostic factors | Risk groups |
| 1.- Time from diagnosis to commencement of treatment less than one year |
|
| 2.- Karnofsky < 80% | |
| 3.- Hemoglobin below the normal lower limit (normal: 12 g/dl) | |
| 4.- Calcium above the upper normal limit (normal: 8,5-10,2 mg/dl) | |
| 5.- Neutrophils above the upper normal limit (normal: 2,0-7,0 x 103/l) | |
| 6.- Platelets above the upper normal limit (normal: 150 000-400 000) |
The patient starts pazopanib at 400 mg/day for three months. A tomographic control of thorax, abdomen, and pelvis is performed, and it is observed that mediastinal metastatic lesions persist, associating chemotherapy with cisplatin 75 mg/m2 scheme in D1 + gemcitabine 1000 mg/m2 in D1 and D8, every twenty-eight days for a total of six cycles. A partial response is achieved at the level of mediastinal ganglion conglomerate.
Eight months after diagnosis, the patient underwent partial mediastinal node metastasectomy (due to para-esophageal presentation), whose pathological study confirmed metastasis of Xp11.2 translocation renal cell carcinoma.
Due to the persistence of the residual para-esophageal ganglion mass, the start of sunitinib is prescribed, a drug that the parents refuse to administer. Therefore, the patient continues with periodic clinical and imaging controls where the stable persistence of this lesion and a Zubrod score of 0 are evidenced.
Up to the time of publication, the patient has a disease-free survival of approximately eighteen months, with periodic tomography checks to assess disease progression.
DISCUSSION
Renal cell carcinomas are very rare childhood neoplasms, accounting for only 2% to 3% of malignant kidney tumors in children. However, its incidence has been shown to increase with age(1).
According to the Japanese Society of Pediatric Surgeons, renal cell carcinomas account for 1.4% of all renal tumors in children under 4 years of age, 15.2% in patients between 5 and 9 years of age, and 52.6% in patients between 10 and 15 years of age, with peak incidence at 9 and 15 years of age(2).
The new category of MiT family translocation renal cell carcinoma has been included into the WHO classification in 2016. The MiT family translocation renal cell carcinoma includes Xp11 translocation renal cell carcinoma and t (6; 11) renal cell carcinoma
It is essential to establish that t-RCC subtypes Xp11 and t (6; 11) are caused by rearrangements in the genes of the transcription factors TFE3 and TFEB, respectively(3,4).
Determining the exact frequency of t-RCC is a challenge. Some authors such as Kmetec et al.(5) have determined that Xp11 t-RCC represent 20-40% of RCC in children, while the data provided by Komai et al.(6)states that approximately one-third of RCC in children will be t-RCC. The rarity of the tumor causes this difference, such that the clinical-pathological characteristics, prognosis, and treatment have been obtained from retrospective data.
The diagnosis of renal cell carcinomas is usually determined by morphology, while the diagnosis of renal cell carcinomas with translocation requires the verification of rearrangements in the transcription factors TFE3 and TFEB, for which the cytogenetic technique of chromosome marking is generally used, whereby these are hybridized with probes that emit fluorescence (FISH)(7,8).
The case of an 11-year-old girl, with a diagnosis of renal cell carcinoma (clinical stage IV as a result of distant lymph node involvement), was presented. She underwent right-sided radical nephrectomy and regional lymphadenectomy, and subsequently initiated a 400 mg/day pazopanib treatment associated with platinum-based chemotherapy (75 mg/m2 cisplatin in D1 + Gemcitabine 1000 mg/m2 in D1 and D8) every 28 days for a total of six cycles, allowing the patient to undergo mediastinal nodal resection and maintain a progression-free survival of approximately 18 months.
Sukov et al. published the results of 632 patients with RCC, of which only six were shown to be carriers of rearrangements of TFE3. The researchers established the disease’s debut imply nodal involvement associated with a disease of aggressive behavior. Also, the study highlighted a worse progression-free survival (PFS) for those patients who carried the TFE3 rearrangement compared to those who were not carriers(9,10).
The overall survival reported for children with RCC varies according to the following factors: 1) the extent of surgery (radical nephrectomy +/- regional lymph node dissection), 2) clinical stage at debut, 3) availability of first-line drugs in metastatic disease and 4) strict patient follow-up. Indolfi P. et al. performed a retrospective analysis of 41 patients, among children and adolescents, with which they demonstrated that patients with RCC have, on average, an overall survival rate of 63%, with survival rates determined by clinical stage. Besides, they pointed out that between stages I and IV, there is a survival rate of 92.4%, 84.6%, 72.7%, and 13.9%, respectively(11).
Overall survival and progression-free survival in children with Xp11.2 translocation renal cell carcinoma are challenging to determine with accuracy because there is no prospective study in this population group, making it difficult to compare with other subtypes of renal cell carcinoma. It has been established that the prognosis of Xp11.2 translocation renal cell carcinomas in children is better compared to that of adults(5,12).
Al-Daghmin et al. performed a retrospective review of 23 patients with Xp 11.2 translocation renal cell carcinoma, including only two patients younger than 18 years (two girls aged 13 and two years, respectively), and concluded that, after a follow-up of 35 months, the rate of disease-free survival in 3 years was 75%(13).
The patient was treated as established by international guidelines and obtained a progression-free survival of approximately 18 months to the present. All this allows us to establish that an adequate initial interdisciplinary management will achieve a long disease-free survival in these patients.
CONCLUSION
Multidisciplinary management from the outset associated with the availability of first-line drugs for this type of neoplasms significantly improves progression-free survival in children with translocation renal cell carcinoma.
Authors' contributions: The authors participated in the genesis of the idea, project design, data collection and interpretation, analysis of results, preparation of the manuscript.
Financing: Self-financed.
Conflict of interest: The authors declare no conflict of interest.
Received: October 10, 2019
Approved: December 13, 2019
Correspondence: Gloria Paredes Guerra
Address: Av Garcilaso de la Vega 1420, Cercado de Lima.
Telephone: +51 981 291 940
Mail: gloriaparedes200@yahoo.com
BIBLIOGRAPHIC REFERENCES
