REPORTE DE CASO
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2021 - Universidad Ricardo PalmaDOI 10.25176/RFMH.v21i1.3488
MALFORMACIÓN CONGÉNITA DE LA VÍA AÉREA PULMONAR (MCVAP), REPORTE DE CASO
CONGENITAL MALFORMATION OF THE PULMONARY AIRWAY (MCVAP), CASE REPORT
José Luis Medina Valdivia1,2
José Luis Medina Valdivia1,2
1Servicio de Pediatría Hospital Regional de Moquegua, Moquegua-Perú.
2Médico Pediatra.
Las malformaciones pulmonares comprenden distintas anomalías del sistema respiratorio, entre ellas la malformación congénita de la vía aérea pulmonar (MCVAP), antes conocida como malformación adenomatosa quística, que es una enfermedad rara con una incidencia de 1 por 8 300 a 35 000 nacidos vivos. Se ha descrito cinco patrones de clasificación de acuerdo al número y tamaño de quiste, además de sus características histológicas, siendo la MCVAP tipo 1 la más frecuente, presenta desplazamiento de estructuras adyacentes según tamaño, asociados a carcinoma broquioalveolar, y de buen pronóstico tras resección quirúrgica. Presentamos el caso, de una paciente de sexo femenino de cuatro años de edad con hospitalizaciones recurrentes por neumonía y síndrome obstructivo bronquial. La acuciosa anamnesis y examen físico complementando con la radiografía de tórax y tomografía permitió la sospecha diagnóstica. Posteriormente la paciente fue intervenida quirúrgicamente, no se presentaron complicaciones y los síntomas respiratorios desaparecieron. El estudio histopatológico confirmó el diagnóstico.
Palabras Clave: Enfermedad pulmonar; anomalías congénitas; quistes; neumonía; niño hospitalizado (fuente: DeCS BIREME).
ABSTRACT
Pulmonary malformations include different abnormalities of the respiratory system, including congenital pulmonary airway malformation (MCVAP), formerly known as cystic adenomatous malformation, which is a rare disease with an incidence of 1 in 8,300 to 35,000 live births. Five classification patterns have been described according to the number and size of the cyst, in addition to their histological characteristics, with type 1 MCVAP being the most frequent, showing displacement of adjacent structures according to size, associated with brochioalveolar carcinoma, and good prognosis after resection surgical. We present the case of a four-year-old female patient with recurrent hospitalizations for pneumonia and bronchial obstructive syndrome. The thorough anamnesis and physical examination supplemented with the chest x-ray and tomography allowed the diagnosis to be suspected. Later, the patient underwent surgery, there were no complications and the respiratory symptoms disappeared. The histopathological study confirmed the diagnosis.
Key words: Pulmonary disease; congenital anomalies; cysts; pneumonia; hospitalized child (source: MeSH NLM).
Las anomalías congénitas del aparato respiratorio comprenden un extenso número de patologías que pueden comprometer el desarrollo de la laringe, la tráquea y los bronquios, el parénquima pulmonar, el diafragma o la pared torácica. Las malformaciones congénitas pulmonares se dividen en: malformaciones congénitas del tracto respiratorio superior e inferior; entre estas últimas se encuentra la malformación congénita de la vía aérea pulmonar (MCVAP); anteriormente conocida como malformación adenomatoidea quística congénita, secuestro broncopulmonar, sobreinsuflación lobular congénita y atresia bronquial (1).
La MCVAP es una enfermedad rara con una incidencia de 1 por 8 300 a 35 000 nacidos vivos. Representa aproximadamente el 25% de todas las lesiones congénitas del pulmón, tiene igual frecuencia en el pulmón derecho que en el izquierdo y ligero predominio en varones. La enfermedad unilobar es mucho más común (85–95%) que la multilocular (1,2).
La MCVAP se puede presentar al nacimiento como un síndrome incompatible con la vida; los afectados con frecuencia son prematuros, mortinatos o neonatos que fallecen rápidamente por insuficiencia respiratoria o anasarca. Únicamente el 10% de los casos se manifiestan en el primer año de vida .Existen casos descritos de niños más grandes, e incluso de adultos, pero son muy infrecuentes, y en general se sospecha por infecciones recurrentes (2).
Se presenta el caso de una niña de cuatro años edad que ingresa al Servicio de Pediatría del Hospital Regional de Moquegua con antecedentes de hospitalizaciones recurrentes de infecciones respiratorias bajas; a quien mediante una cuidadosa anamnesis, un buen examen físico y el apoyo de exámenes complementarios como la radiografía y la tomografía de tórax se plantea la sospecha diagnóstica de MCVAP tipo I que posteriormente se confirma con el estudio histopatológico.
REPORTE DE CASO
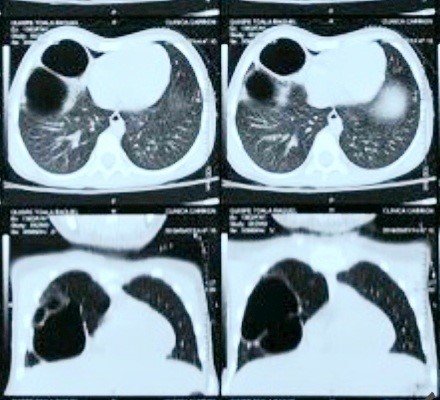
Paciente de cuatro años de edad, sexo femenino, ingresa al Servicio de Pediatría del Hospital Regional de Moquegua por presentar cinco días antes: tos seca, dificultad respiratoria y fiebre; sin antecedentes de importancia al nacer, pero con múltiples hospitalizaciones desde el primer mes de vida por bronquiolitis, síndrome obstructivo bronquial, neumonía recurrente (todas sin complicaciones), y por presunciones diagnósticas de tuberculosis pulmonar. Paciente cursa con evolución estacionaria durante los dos primeros días de hospitalización; luego presenta desaturación de oxigeno hasta 87%, frecuencia respiratoria de 60 respiraciones por minuto, frecuencia cardiaca 100 latidos por minuto y temperatura 37°C. Al examen físico presenta aleteo nasal, asimetría en tórax anterior derecho (Figura 1), tiraje subcostal e intercostal, a la auscultación el murmullo vesicular en hemitórax derecho se encuentra disminuido. Respecto a los exámenes de apoyo al diagnóstico hemograma con leucocitosis, neutrofilia, PCR reactivo (+++), las imágenes en la radiografía y tomografía de tórax (Figuras 2 y 3 respectivamente), evidencian imagen quística bilobulada en el pulmón derecho acompañada de atelectasia basal derecha sugestiva de una malformación pulmonar congénita. Paciente recibe oxígeno, antibiótico y fisioterapia respiratoria. Una vez estable fue referida al Instituto Nacional de Salud del Niño San Borja en donde fue intervenida quirúrgicamente (lobectomía superior derecha y media) confirmando la sospecha diagnóstica histopatológicamente (MCVAP Tipo I: dos cavidades quísticas, la mayor de 7x4x4 cm y la menor 4x3x3 de diámetro, acompañado la ectomía del lóbulo superior y medio de bronquiectasia focal, atelectasia, congestión vascular difusa, bronquiolitis nodular focal, y presencia de macrófagos espumosos y reacción gigantocelular del epitelio del quiste).

|

|
|
Las MCVAP comprenden un espectro de quistes de diferentes tamaños de histología variable. La antigua clasificación de Stocker y colaboradores en el año 1977 incluían 3 tipos, pero en la actualidad contempla 5 grupos según el número y tamaño de quistes, así como origen histopatológico (Tabla 1) (2,3).
Tabla 1. Nueva clasificación de las MCVAP por Stocker y colaboradores.
| Tipo 0 | Disgenesia acinar y displasia de la gran vía aérea, incompatible con la vida |
| Tipo 1 | Quiste único o múltiple de más de 2 cm de diámetro, de bronquio o bronquiolo |
| Tipo 2 | Quiste único o múltiple de menos de 2 cm de diámetro de bronquíolo |
| Tipo 3 | Lesión solida con algún quiste de menos de 0,5 cm de bronquiolo y conducto alveolar |
| Tipo 4 | Numerosos quistes de origen acinar |
El tipo I es el más frecuente de las MCVAP y tiene el mejor pronóstico, debido a que son localizadas y solo afectan a una parte de un lóbulo, también puede ser multiloculados (4). La mayoría está presente en el periodo neonatal o en el intrauterino, pero en casos infrecuentes puede presentarse más tarde. La presentación clínica es variable y depende directamente del tamaño de la masa pulmonar; en la vida prenatal el peor pronóstico y alta mortalidad se asocia al desarrollo de hidrops fetal; en la vida post natal el cuadro clínico es variable y está igualmente relacionado con el tamaño de la lesión pulmonar(5).Microscópicamente existe un límite marcado entre la lesión y el pulmón normal adyacente, pero no existe cápsula. Los espacios quísticos de mayor tamaño están tapizados por el epitelio columnar seudoestratificado ciliado y el pulmón entre los quistes puede mostrar sobrecrecimiento y parénquima alveolar poco desarrollado. Históricamente en el 35- 50% de los casos se describe hiperplasia de células mucosas. La transformación maligna es infrecuente, pero las áreas de consolidación que no se resuelven durante el seguimiento por imágenes de las MCVAP deberían preocupar (adenocarcinoma mucinoso). El pronóstico es muy bueno tras la resección completa, aunque se ha descrito recurrencia de la enfermedad si se reseca de forma incompleta, en ocasiones décadas después de la resección original del quiste. También puede mostrar indicios de infección sobreañadida cuando son resecadas, incluso un aspergiolioma.
El diagnóstico de las malformaciones congénitas de la vía área pulmonar puede resultar difícil en ciertas oportunidades, puede imitar clínica y/o radiológicamente otras enfermedades; es conveniente que se encuentre presente en el pensamiento médico para facilitar su diagnóstico precoz (6).
La resección quirúrgica está indicada para evitar complicaciones pulmonares futuras como son infección recurrente, compresión de estructuras adyacentes y transformación maligna (7). Se ha reportado en la literatura la escisión toracoscópica aparentemente con buenos resultados (8). El diagnóstico definitivo se establece mediante el estudio histopatológico al demostrarse que la pared del quiste esta revestida por epitelio bronquial (8,10). En conclusión, el diagnóstico prenatal de las malformaciones pulmonares congénitas de la vía pulmonar permite corroborar el diagnóstico postnatal tempranamente, lo que ayuda a tomar acciones terapéuticas oportunas para prevenir complicaciones posteriores. Ésta patología es diagnosticada muy infrecuentemente en lactantes, preescolares, escolares y/o adolescentes; el tratamiento quirúrgico temprano evita posibles complicaciones pulmonares futuras.
Guzmán et al. (11) reportan un caso similar de una niña de 4 años de edad en Colombia con infecciones respiratorias recurrentes incluso recibió tratamiento para tuberculosis. En el año 2018 Ventura et al. (12) en Instituto Nacional Materno Perinatal de la ciudad de Lima-Perú reportan los primeros tres casos de malformación congénita de la vía área pulmonar con diagnóstico prenatal tratados intrauterinamente. Wang (13) en el año 2019, en Argentina, reporta un caso de MCVAP gigante que imita un neumotórax a tensión (13).
Presentamos un caso Servicio de Pediatría del Hospital Regional de Moquegua, de MCVAP Tipo I, una enfermedad congénita que en nuestro medio no es diagnosticada frecuentemente, quizás por la dificultad de realizar ecografías gestacionales de alta resolución. La paciente con antecedentes de infecciones respiratorias frecuentes y que tuvo que esperar hasta los cuatro años de edad para ser diagnosticada y tratada.
CONCLUSIÓN
En países en desarrollo donde es difícil acceder a sofisticados recursos tecnológicos como ayuda diagnóstica, el realizar una historia clínica detallada y completa, como en el caso de esta paciente, permitió observar la recurrencia de hospitalizaciones con diagnósticos respiratorios no concluyentes, plantear los diagnósticos presuntivo y diferencial y referir a la paciente para el tratamiento quirúrgico oportuno.
Contribuciones de Autoría: El autor participó en la concepción y diseño del trabajo; recolección, análisis e interpretación de datos; revisión crítica y redacción de la versión final.
Financiamiento: Autofinanciado.
Conflictos de intereses: El autor declara no tener conflictos de interés en la publicación de este reporte de caso.
Recibido: 13 de diciembre 2020
Aprobado: 13 de diciembre 2020
Correspondencia: José Luis Medina Valdivia
Dirección: Urbanización los Damascos C-1, Moquegua, Mariscal Nieto-Perú
Teléfono: 953951080
Correo: jlzf29@hotmail.com
REFERENCIAS BIBLIOGRÁFICAS
