REPORTE DE CASO
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2021 - Universidad Ricardo PalmaDOI 10.25176/RFMH.v21i1.3488
CONGENITAL PULMONARY AIRWAY MALFORMATION (CPAM): A CASE REPORT
MALFORMACIÓN CONGÉNITA DE LA VÍA AÉREA PULMONAR (MCVAP), REPORTE DE CASO
José Luis Medina Valdivia1,2
1Pediatric Services Hospital Regional de Moquegua, Moquegua-Peru.
2Pediatrician.
Pulmonary malformations include different anomalies of the respiratory system, including congenital pulmonary airway malformation (CPAM), formerly known as cystic adenomatous malformation, which is a rare disease with an incidence of 1 per 8,300 to 35,000 live births. Five classification patterns have been described according to the number and size of the cyst, in addition to their histological characteristics, with type I CPAM being the most frequent, showing displacement of adjacent structures according to size, associated with brochioalveolar carcinoma, and favourable prognosis after surgical resection. We present the case of a four-year-old female patient with recurrent hospitalizations for pneumonia and bronchial obstructive syndrome. The careful anamnesis and physical examination supplemented with the chest X-ray and tomography allowed the diagnosis to be suspected. Later, the patient underwent surgery, there were no complications and the respiratory symptoms disappeared. The histopathological study confirmed the diagnosis.
Key words: Pulmonary disease; congenital anomalies; cysts; pneumonia; hospitalized child (source: MeSH NLM).
RESUMEN
Las malformaciones pulmonares comprenden distintas anomalías del sistema respiratorio, entre ellas la malformación congénita de la vía aérea pulmonar (MCVAP), antes conocida como malformación adenomatosa quística, que es una enfermedad rara con una incidencia de 1 por 8 300 a 35 000 nacidos vivos. Se ha descrito cinco patrones de clasificación de acuerdo al número y tamaño de quiste, además de sus características histológicas, siendo la MCVAP tipo 1 la más frecuente, presenta desplazamiento de estructuras adyacentes según tamaño, asociados a carcinoma broquioalveolar, y de buen pronóstico tras resección quirúrgica. Presentamos el caso, de una paciente de sexo femenino de cuatro años de edad con hospitalizaciones recurrentes por neumonía y síndrome obstructivo bronquial. La acuciosa anamnesis y examen físico complementando con la radiografía de tórax y tomografía permitió la sospecha diagnóstica. Posteriormente la paciente fue intervenida quirúrgicamente, no se presentaron complicaciones y los síntomas respiratorios desaparecieron. El estudio histopatológico confirmó el diagnóstico.
Palabras Clave: Enfermedad pulmonar; anomalías congénitas; quistes; neumonía; niño hospitalizado (fuente: DeCS BIREME).
Congenital anomalies of the respiratory system include a large number of pathologies which can affect the development of the larynx, trachea and bronchi, lung parenchyma, diaphragm and thoracic wall. Congenital pulmonary malformations are divided into: upper and lower respiratory tract congenital malformations. Congenital pulmonary airway malformation (CPAM), formerly known as congenital cystic adenomatous malformation, pulmonary sequestration, congenital lobar emphysema and bronchial atresia (1), appear in the lower respiratory tract congenital malformations.
CPAM is a rare disease with an incidence of 1 per 8,300 to 35,000 live births. It represents approximately 25% of all congenital lung disease, it has equal frequency in the right lung than in the left one and slight predominance in males. Unilobular disease is much more common (85-95%) than multilobular disease (1,2).
CPAM may present at birth as an incompatible with life syndrome. Prematures, stillbirths and neonates are frequently affected. They promptly die from respiratory insufficiency and anasarca. Only 10% of the cases shows up during the first year of life. There are cases described in older children, and even adults; however, those cases are very uncommon and generally suspected of recurrent infections (2).
A case of 4-year-old girl who was admitted to the Pediatric Services of Hospital Regional de Moquegua with history of repeated hospitalizations because of lower respiratory infections and suspected diagnosis of congenital pulmonary airway malformation type I is presented. This is due to a careful anamnesis, good physical examination, auxiliary imaging examinations such as X-ray and tomography, with a subsequent diagnostic confirmation through histopathological study.
CASE REPORT
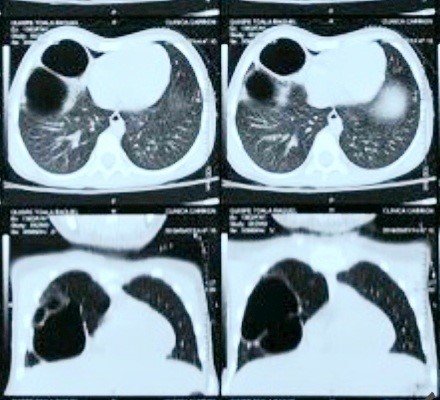
A 4-year-old female patient was admitted to the Pediatric Services of Hospital Regional de Moquegua because of dry cough, respiratory distress and fever the previously 5 days. No significant history at birth, but she was hospitalized several times because of bronchiolitis, bronchial obstructive syndrome, recurrent pneumonia (all of them without complications) and presumptive diagnosis of pulmonary tuberculosis since the first month of life. The patient showed stable progression during the first two days of hospitalization. Thereafter, she presented oxygen desaturation up to 87%, heart rate of 100/minute and temperature of 37°C. On physical examination, nasal flaring, asymmetry of anterior right chest ( Figure 1), subcostal and intercostal retractions were detected. On auscultation, vesicular sound in right hemithorax was decreased. Complementary examination related to the complete blood count with leukocytosis, neutrophilia, CRP (+++), the images of the chest radiograph and chest CT (Figure 2 and 3 respectively) revealed bilobed cystic image in right lung coupled with basal right atelectasis suggestive of a congenital pulmonary malformation. Patient received oxygen, antibiotic and respiratory physiotherapy. After stable condition, she was referred to Instituto Nacional de Salud del Niño San Borja where she underwent surgery (right upper and middle lobectomy) confirming the diagnostic suspicion based on histopathological study (CPAM type I: two cystic cavities, the biggest 7x4x4 cm in size and the smallest 4x3x3 in diameter, along with upper and middle lobe ectomy of focal Bronchiectasis, atelectasis, diffuse vascular congestion, focal nodular bronchiolitis, and presence of foamy macrophages and foreign-body giant cell reaction of the cyst epithelium).

|

|
|
CPAM comprise a cysts range with various sizes of variable histology. In 1977, the early Stocker et al.’s classification included three types. However, it is now classified into five groups, based on the cysts amount and size, as well as their histopathological origin (Table 1) (2,3).
Table 1. New Stocker et al.’s classification of CPAM.
| Type 0 | Acinar dysgenesis and dysplasia of the airways. Incompatible with life |
| Type 1 | One or multiple cysts over 2 cm in diameter of bronchus or bronchiole |
| Type 2 | One or multiple cysts under 2 cm in diameter of bronchiole |
| Type 3 | Solid lesion with some cyst under 0,5 cm of bronchiole and alveolar duct |
| Type 4 | Acinar origin multiple cysts |
Among CPAM Type I, is the most common. The fact of lesions being located and can only affect a part of one lobe makes it the type with the best prognosis. It can also be multilocated (4). Most of the cases occur in the neonatal period or intrauterine period, but in rare cases they may show later. The clinical presentation is variable and relies directly on the size of the lung mass. Hydrops fetalis development is associated with the worst prognosis and high mortality during prenatal stage. Clinical picture is variable and is related to the size lung lesion too (5). Microscopically, there is a marked boundary between the lesion and the adjacent normal lung, but there is no capsule. The larger cystic spaces are covered by the pseudostratified ciliated columnar epithelium and the lung between the cysts may reveal overgrowth and underdeveloped alveolar parenchyma. Historically, mucous cell hyperplasia is described in 35 to 50% of the cases. Malignant transformation is rare, but areas of consolidation that are not resolved during CPAM imaging should be considered (mucinous adenocarcinoma). After complete resection the prognosis is favourable, although recurrence of the disease has been described if it is completely resected, sometimes decades after the original cyst resection. It may also show signs of additional infection when resected, including an aspergilloma.
Sometimes the diagnosis of congenital pulmonary airway malformations can be difficult. Whether clinically or radiologically it can simulate other diseases. Therefore, health care providers should be aware of such fact in order to furnish an early diagnosis (6).
Surgical resection is intended to prevent further lung complications such as recurrent infection, compression of adjacent structures and malignant transformation (7). Thoracoscopic excision has been suggested in literature of having apparently good results (8). The definitive diagnosis is determined by histopathological study as it is showed that cyst wall is covered by the bronchial epithelium (8,10). In conclusion, prenatal diagnosis of the congenital malformation of the pulmonary tract allows the earlier postnatal diagnosis confirmation. This contributes in taking timely therapeutic action to prevent further complications. Uncommonly this pathology is diagnosed in infants, preschoolers, school children and/or adolescents. Early surgical treatment prevents from possible future pulmonary complications.
In Colombia, a similar case of a 4-year-old girl who presented recurrent respiratory infections and even received treatment for tuberculosis was reported by Guzmán et al. (11). In 2018, Ventura et al. (12) reported the first three cases of congenital pulmonary airway malformation with prenatal diagnosis intrauterinally treated at Instituto Nacional Materno Perinatal located in Lima, Peru. In Argentina a huge CPAM case that simulates tension pneumothorax (13) was reported by Wang (13) in 2019.
We reported a case of CPAM Type I at the Pediatric Services of Hospital Regional de Moquegua. This is a rarely diagnosed disease in our environment, perhaps due to the difficulty of carrying out high resolution gestational ultrasounds.The patient, with a history of frequent respiratory infections, had to wait until she was four years old to be diagnosed and treated.
CONCLUSION
In developing countries where it is difficult to access sophisticated technological resources as a diagnostic aid, taking a detailed and complete medical history, as in the case of this patient, made it possible to observe the recurrence of hospitalizations with inconclusive respiratory diagnosis, to propose presumptive and differential diagnosis as well as to refer the patient for timely surgical treatment.
Author contributions: The author participated in the conception and design of the study, data collection, analysis and interpretation, critical review and writing of the final version.
Financing: Self-financing
Conflict of interest: The author declares that he has no conflict of interest in the publication of this case report.
Received: December 13, 2020
Approved: December 13, 2020
Correspondence: José Luis Medina Valdivia
Address: Urbanización los Damascos C-1, Moquegua, Mariscal Nieto-Perú
Mobile: 953951080
Email: jlzf29@hotmail.com
REFERENCES
