ARTICULO REVISIÓN
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2021 - Universidad Ricardo Palma10.25176/RFMH.v21i2.3626
TRATAMIENTO DE LAS ENFERMEDADES GENÉTICAS: PRESENTE Y FUTURO
MANAGEMENT OF GENETIC DISEASES: PRESENT AND FUTURE
Hugo Hernán Abarca Barriga (1,2,3,a,b), Milana Trubnykova (2,a), María del Carmen Castro Mujica (1,a)
1 Facultad de Medicina Humana, Universidad Ricardo Palma, Lima-Perú.
2 Servicio de Genética & EIM, Instituto Nacional de Salud del Niño-Breña, Lima-Perú.
3 Universidad Científica del Sur, Lima-Perú
a MD Especialista en Genética médica
b Magister en Genética
RESUMEN
El número de enfermedades genéticas se estima que podrían ser más de 10 000 condiciones diferentes, afectando alrededor del 6-8% de la población. La presente revisión nos muestra la importancia del descubrimiento de las variantes patogénicas en nuestro genoma que nos permite conocer con mayor precisión cuales son los mecanismos fisiopatológicos y, por lo tanto, conocer puntos dianas susceptibles de modificaciones mediante diferentes estrategias terapéuticas para poder palear los síntomas y signos, aumentar la expectativa de vida, mejorando así la calidad de vida de los pacientes que tienen algunas de estas enfermedades genéticas. Las diferentes terapias que existen en la actualidad son muy diversas como fármacos de uso en patologías comunes, terapia nutricional, fórmulas especiales, terapias de reemplazo enzimático, trasplante de órganos y células hematopoyéticas, reducción de sustrato, oligonucleótidos y la terapia génica. Al ser las enfermedades genéticas clínicamente heterogéneas, abre la posibilidad de poder investigar cada vez más nuevas estrategias en un mayor número de enfermedades que en la actualidad están olvidadas.
Palabras Clave: Enfermedades genética, terapia genética, Células Madre hematopoyéticas, trasplantes, terapias (Fuente: DeCS BIREME)
ABSTRACT
Today, the number of genetic diseases is around 10000 conditions, affecting to 6%-8% of all populations. This review shows us how the discovery of genetic variants in our genome, this facilitated to know with precision about the mechanisms physiopathological, and hence to recognize those target points susceptible to modifications, through therapeutical strategies different with palliative proposals, increase life expectancy, or improve qualities of life. These therapies are diverse, using drugs for polygenic diseases, nutritional therapy, special formulas, enzyme replacement therapies, hematopoietic stem cell transplant, substrate reduction, oligonucleotides, and gene therapy. These genetic diseases are heterogeneous clinically with a very low frequency; nevertheless, open to the possibility of research in new strategies for more genetic disease, that today, furthermore, are orphans.
Keywords: Genetic diseases; genetic Therapy, hematopoietic Stem Cells, transplant, therapy (Source: MeSH NLM)
INTRODUCCIÓN
Una enfermedad rara está definida por la frecuencia de aparición. Es así por ejemplo que, en
Europa refieren es aquella con una incidencia menor a 1/2 000 personas. El número de pacientes afectados
se estima entre el 6-8% de la población en general. En nuestro País no existen estudios que definen el
número real de personas afectadas estimándose por lo tanto que son aproximadamente más de 2 millones de
peruanos(1). Sin embargo, se hicieron algunos estudios que estiman que el
porcentaje de afectados por una enfermedad rara estaría entre el 3,5-5,9%(2).
La etiología de las enfermedades raras es de origen genético en un 80% de los casos, y el 20%
restante, de origen desconocido. Las de origen genético las podemos dividir en tres grupos: i) las que
se producen por variantes en único nucleótido (SNV, del inglés nucleotide variant), ii) variantes de
múltiples nucleótidos (MNV, del inglés multinucleotide variant) y iii) variantes en el número de copias
(CNV, del inglés copy number variation). Las dos primeras variantes, principalmente, producen las
enfermedades monogénicas, las que se estiman, según la Organización Mundial de la Salud (OMS), que son
más de 10 000 entidades(1). Las CNV patogénicas (o probablemente patogénicas)
que provocan síndromes de microdeleción / microduplicación; donde las más frecuentes tienen una
prevalencia entre 1/1 000 a 1/25 000(3); aunque, se ha reportado que en fetos
la incidencia de las CNV es más alta llegando al 0,7%(4). Es importante
aclarar que no todas las enfermedades genéticas son raras. (ej. síndrome Down, síndrome Klinefelter)(
5). De todo este gran grupo de condiciones, alrededor de 500 enfermedades tienen un tratamiento
dirigido(6).
Se tiene que resaltar que las enfermedades genéticas representan hasta el 71% de las
hospitalizaciones pediátricas(7), y provocan entre el 20 y el 30% de muertes
de este grupo etario(8). Esta proporción de pacientes genera un gran impacto
económico en los sistemas de salud; es así que, un estudio australiano desarrollado en una cohorte
poblacional durante el año 2010, encontró que los pacientes con enfermedades raras generaron el 10,5% de
gastos hospitalarios(9), además de una mayor permanencia hospitalaria que sus
pares sin condiciones genéticas(7).
Las manifestaciones clínicas de las enfermedades genéticas son muy diversas, es decir tienen una
gran variabilidad clínica o fenotípica y pueden manifestarse como hipotonía, retraso del desarrollo
psicomotor, discapacidad intelectual, epilepsia, neuroregresión, anomalías congénitas, talla baja,
microcefalia, inmunodeficiencias primarias, esquizofrenia, trastornos del espectro autista, trastornos
de conducta, déficit de atención e hiperactividad, demencia, movimientos anormales y cáncer. Incluso,
hay entidades, como la parálisis cerebral infantil, en las que anteriormente no se describía un
componente genético y ahora se considera que hasta un 20% de los casos tiene una causa genética(1). Es importante precisar que las enfermedades genéticas pueden aparecer en
cualquier etapa de la vida, desde prenatal hasta la adultez(10).
Desde fines del siglo XX, gracias a la decodificación del ADN y el mejor entendimiento de la
fisiopatología de las enfermedades genéticas, las terapias específicas, llámese aquellas que son
dirigidas al factor o factores que inician la enfermedad, se han ido incrementando de manera progresiva
y sostenida, el cual es coadyuvado a través de la bioinformática(11).
Existen terapias para las enfermedades genéticas que están disponibles y que su uso ha sido
aprobado por instituciones internacionales como la Food and Drug Administration (FDA)(12,12) y European Medicine Agency (EMA)(14,15). Por otro lado, existe una gran expectativa de
tratamientos nuevos los cuales están en investigación básica y algunos de ellos en investigación
clínica, tal como se puede apreciar en el portal del clinical trials observando más de 2 520 diferentes
estudios(16).
En esta revisión tenemos como objetivo intentar identificar de manera general el tratamiento
farmacológico existente en la actualidad y qué se está investigando en estas enfermedades genétocas. La
manera como se realiza una aproximación terapéutica está enfocada en alguno de los puntos de la cascada
fisiopatológica de las enfermedades genéticas. Es así, por lo tanto, que el tratamiento podría ser a
nivel del gen(es) afectado(s) (ej. terapia génica y cromosómica), sustituyendo la proteína anómala (ej.
trasplante de células hematopoyéticas), modificando la cascada metabólica (ej. formulas especiales,
terapia de reducción de sustrato) y el sintomático(17) (Figura 1)
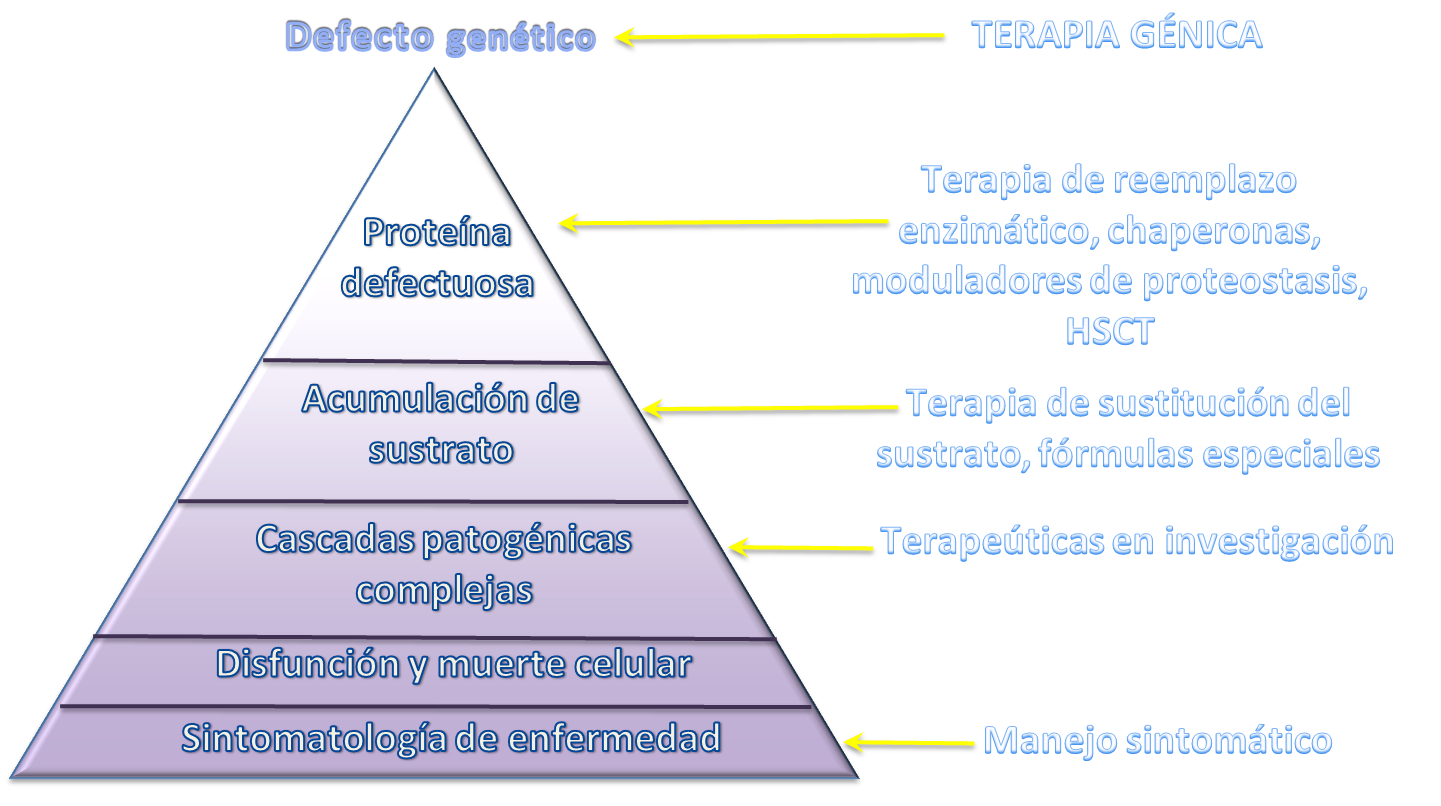
|
1. TERAPIA GÉNICA
El objetivo principal de la terapia génica (Conocida también como genoterapia) es incorporar de
manera suficiente una expresión duradera de un gen o transgen terapéutico con la finalidad de mejorar o
curar los síntomas con eventos adversos mínimos(18).
Cuando se iniciaron las investigaciones se enfocó principalmente en aquellas enfermedades
monogénicas. Sin embargo, actualmente la mayoría de los estudios clínicos de investigación están
dirigidos a cáncer(19) (Figura 2).
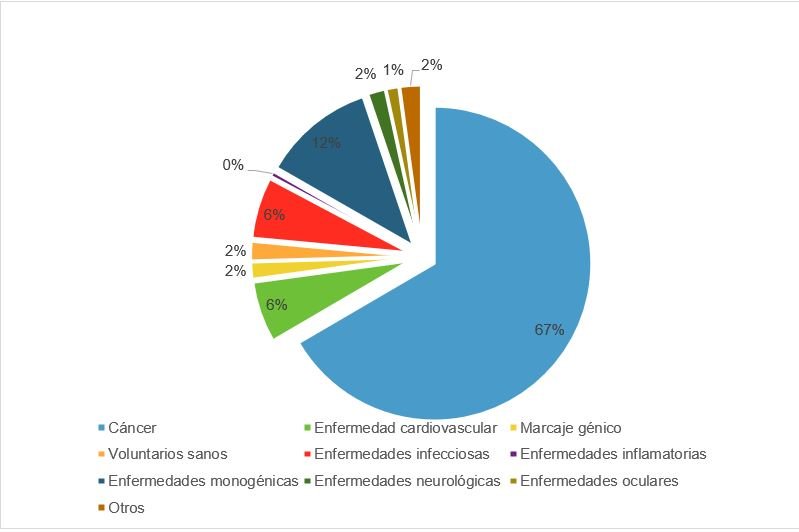
|
Los tipos de genoterapias están dirigidas a células germinales (espermatozoides u óvulos) o a
las células somáticas. La duración de la expresión del gen transferido es según el tipo de patología,
por ejemplo, en las enfermedades monogénicas el tiempo deberá ser prolongado, mientras que en aquellas
enfermedades multifactoriales (ej. cáncer, enfermedades infecciosas) serían de un tiempo corto(20).
Las formas de transferencia génica son de dos tipos: in-vivo y ex-vivo(21). La primera significa que se realiza la entrega directamente a un tejido;
mientras que en la segunda forma se extraen las células del paciente, se realiza la entrega del gen y
luego se vuelve incorporar al individuo afectado(18,21). Los
tipos de terapia génica los podemos subdividir en las que utilizan terapia mediada por virus y
nanopartículas, nucleótidos cortos sintéticos, así como la edición génica(22).
2. TERAPIAS BASADAS EN VIRUS.
Los virus que se usan con más frecuencia son: adenovirus, virus adeno-asociados, lentivirus, retrovirus(23,24)(Figura 3). Los virus adenoasociados son los que tienen más uso por que tienen una capacidad mayor de infección a diferentes tejidos y con una menor respuesta inflamatoria(20).
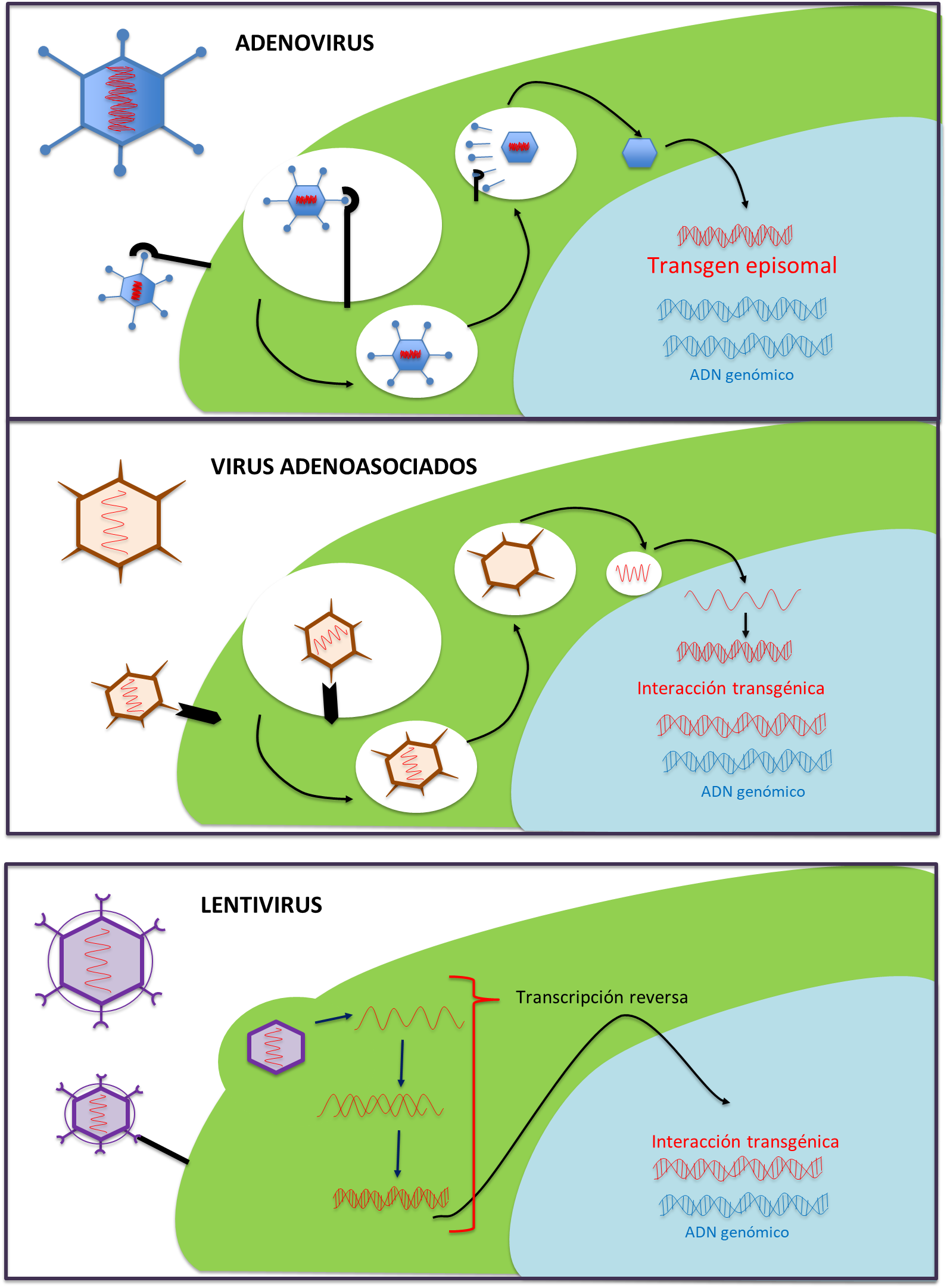
|
Desde el año 2016 a la fecha se han aprobado (por FDA y EMA) genoterapias basadas en virus, las cuales mencionamos a continuación(18,25):
- Alipogene tiparvovec -Glybera- es un virus adeno-asociado (AAV1) que se utiliza para la hiperlipoproteinemia tipo 1 (MIM #238600) ocasionado por variantes recesivas del gen LPL, provocando una deficiencia de lipoprotein-lipasa, causando hiperquilomicronemia y pancreatitis(26).
- Strimvelis, utiliza como vector un retrovirus, que se utiliza en la deficiencia de adenosina desaminasa (gen ADA), que se caracteriza por una inmunodeficiencia combinada severa (MIM #102700)(27).
- Zynteglo, utiliza como vector un lentivirus, es empleado en la betatalasemia (MIM #613985), que se caracteriza por anemia microcítica hipocrómica congénita, disminución de hemoglobina (Hb) A y aumento de Hb F, hepatoesplenomegalia(28).
- Voretigene neparvovec-rzyl -Luxturna- (AAV2), aprobado para el uso de variantes recesivas del gen RPE65 que provoca amaurosis congénita Leber (MIM #204100) y retinitis pigmentosa 20 (MIM #613794)(29).
- Onasemnogene abeparvovec-xioi -Zolgensma- (AAV9), que se utiliza para atrofia muscular espinal 1 (MIM #253300), quienes son niños que presentan hipotonía congénita progresiva, donde la mayoría de los afectados (95-98%) presentan una deleción en el exón 7 del gen SMN1(30).
3. TERAPIAS CON NUCLÉOTIDOS CORTOS
Dentro de las terapias que usan nucleótidos sintéticos cortos, se tiene dos tipos:
- Oligonucleótidos antisentido (AON, del inglés antisense oligonucleotide), tienen 20-30 nucleótidos de ADN, donde las formas de acción son dos: i) utilizando la ARN Hasa, en la cual destruye el ARN mensajero (ARNm) y ii) sin utilizar la ARN Hasa, donde puede actuar modulando el empalme, mediante bloqueo estérico, unión a la región 5’ cap del ARNm o de región 3’ poli A(22,31-33) (Figura 4A).
- ARN de interferencia (ARNi), se utilizan como mecanismo de defensa natural contra los virus ARN. El mecanismo de acción es mediante la utilización de los complejos moleculares Dicer (ribonucleasa) y RISC (del inglés, RNA-induced silencing complex), uniéndose de manera complementaria al ARNm y su posterior rompimiento(22,31-33) (Figura 4B).
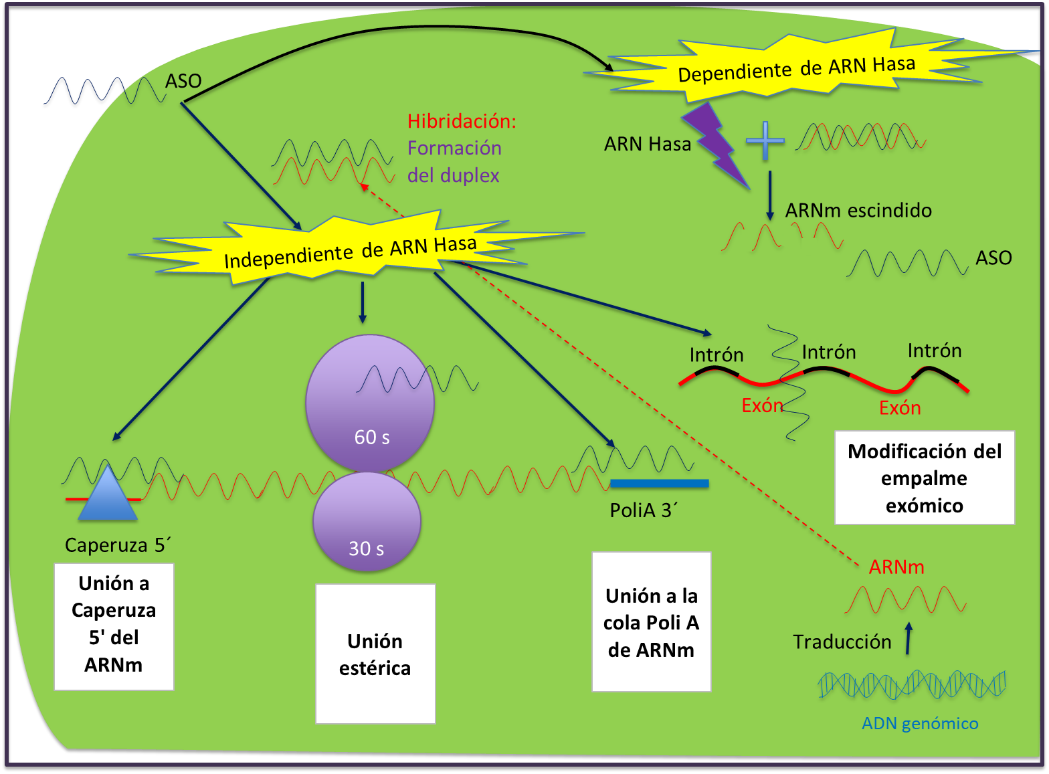 |
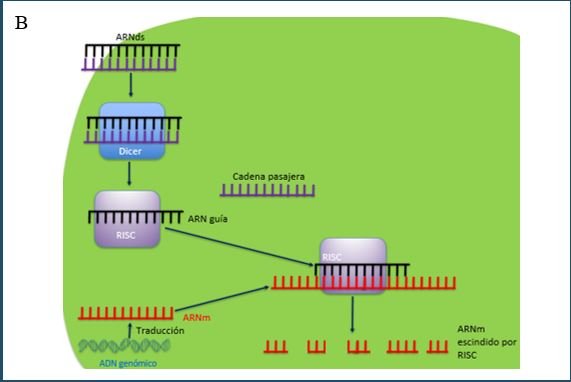 |
Tenemos, a la fecha, aprobadas por FDA y/o EMA las siguientes moléculas:
- Eteplirsen: es un AON que se utiliza en pacientes con distrofia muscular Duchenne (MIM #310200) y que presenten la deleción del exón 51 del gen DMD; haciendo que exista una omisión de este exón provocando una proteína corta, sin embargo, con mayor funcionalidad(34,35).
- Nusinersen: es un AON utilizado en la atrofia muscular espinal tipo 1(MIM #253300), la cual es provocada por variantes en homocigosis del gen SMN1. Este AON es utilizado en pacientes que tienen al menos una copia del gen SMN2, modificando la expresión del gen SMN2 (la cual usualmente se encuentra disminuida), siendo una proteína similar al SMN1(36-38).
- Paitisiran: es un ARNi que se utiliza en la amiloidosis hereditaria relacionada a la transtiretina (MIM #105210), provocada por variantes monoalélicas heterocigotas en el gen TTR. Este ARNi provoca la reducción de la proteína “mutante”(39).
- Mipomersen: es un AON utilizado en hipercolesterolemia familiar (variantes en los genes LDLR, APOB, PCSK9)(40).
4. EDICIÓN GÉNICA/GENÉTICA
Por otro lado, es de suma importancia conocer que se está abriendo mayores posibilidades con el uso de la edición génica a través de meganucleasas, nucleasas como el ZNF (del inglés zinger nucleares finger), TALE (del inglés transcription activator-like repeat) y el CRISPR/Cas9 (del inglés clustered regularly interspaced short palindromic repeat / CRISPR associated protein 9 o en español repeticiones cortas palindrómicas agrupadas e interespaciadas regularmente). Este último sistema, está basado en un sistema que se encuentra en bacterias y archae, el cual confiere resistencia a los virus. El CRISPR/Cas 9 contiene dos elementos, una endonucleasa (Cas 9) y una secuencia simple que sirve como guía (sgARN) (Figura 5A). Los usos que se tiene van desde la regulación génica (Figura 5B-5E), modificación epigenética hasta la imagenología del genoma. Las enfermedades monogénicas que se encuentran en investigación básica son catarata congénita, distrofia muscular Duchenne, tirosinemia hereditaria tipo 1, fibrosis quística, betatalasemia, desórdenes del ciclo de la urea(41).
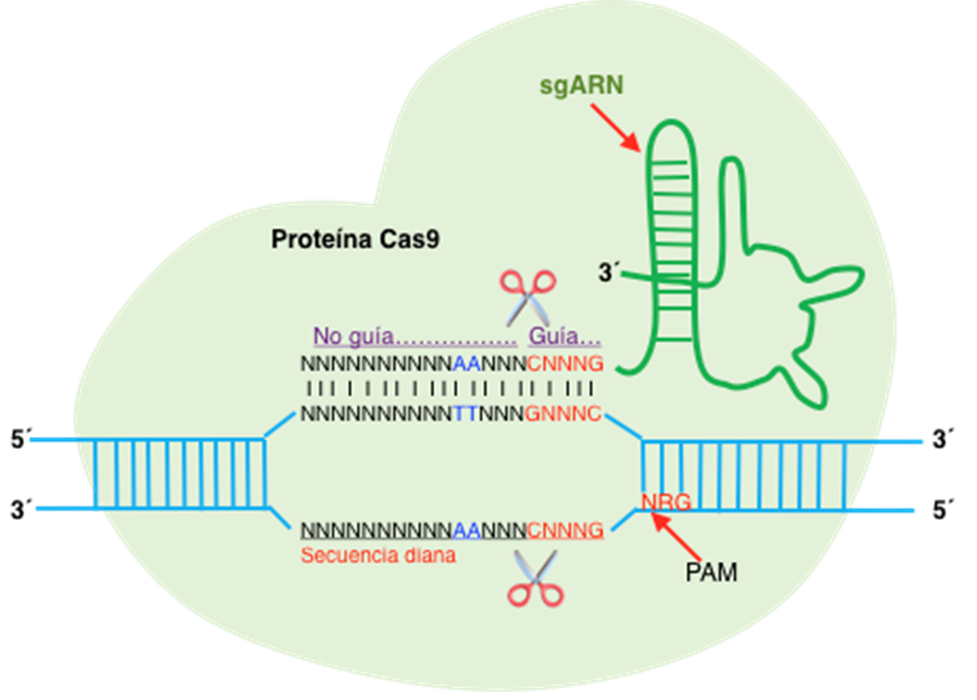 |
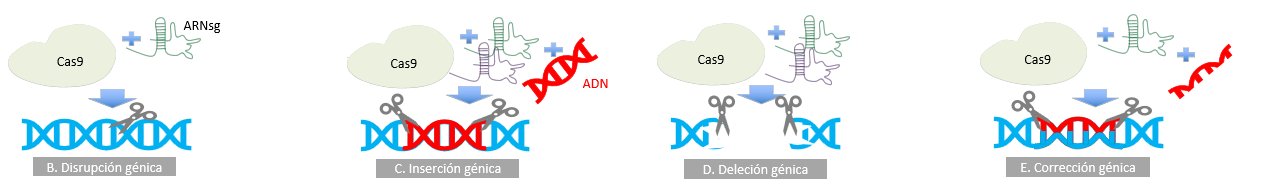 |
5. TERAPIA GÉNICA MEDIANTE VECTORES NO VIRALES
Son estrategias en investigación, que tienen la posibilidad de poder incorporar ADN a través de vectores sintéticos, los cuales frecuentemente son conocidos como nanopartículas (NP) que miden de 10 a 500 nm23. Estas NP tienen la ventaja de síntesis muy fácil, costos de producción menores a los vectores virales, mayor seguridad, capacidad de transportar moléculas más grande y además mayor eficacia(23). Estas nanopartículas pueden ser por ejemplo compuestos por polisacáridos, lípidos sólidos o recubiertos por CK30PEG (del inglés 30-mer cationic polylysine conjugated with 10KDa polyethilene glycol)(23). Por otro lado, se está probando la incorporación del ADN si la utilización de algún vector (ADN “desnudos”) mediante métodos físicos como la electroporación, sonoporación, magnetofección y genes “bala”(20).
6. TERAPIA NUTRICIONAL
Este tipo de terapia está usado principalmente en los errores innatos del metabolismo
(EIM)(42). Es importante recalcar que existen al menos 81 patologías que con
un diagnóstico precoz y un tratamiento oportuno, evitará el riesgo de padecer discapacidad intelectual
(www.treatable-id.org)(43). Es de suma importancia recalcar que el ideal del
momento del diagnóstico es lo más precoz, y si es posible a través del tamizaje neonatal universal de al
menos las entidades más frecuentes(44). Podemos dividir a este tipo de
terapias en(45,46) (Tabla 1):
6a.- Restricción de nutrientes
Al conocer que existe un aumento de un metabolito tóxico por la disminución de la actividad
enzimática, y que existen otros metabolitos cascada arriba; lo que se realiza es disminuir estos a
través de fórmulas especiales, provocando que el tóxico disminuya, evitando así el inicio de la cascada
fisiopatológica(47,48).
6b.- Suplemento nutricional
En muchas oportunidades aparte de la restricción de nutrientes con formulas especiales, se tiene
que suplementar con metabolitos que no se producen de manera adecuada(45,46).
6c.- Eliminación o bloqueo de síntesis del metabolito tóxico
Existen muchos EIM, que la fisiopatología del cuadro se enmarca principalmente en la producción
alternativa de un metabolismo tóxico, por lo que es necesario de drogas o procedimientos (ej. el uso de
hemofiltración en defectos del ciclo de la urea) que eliminen o bloqueen la síntesis de estos(45,46).
Tabla 1. Manejo de algunos errores innatos del metabolismo de aminoácidos, carbohidratos y lípidos.
| ENFERMEDAD | CARACTERÍSTICAS CLÍNICAS SIN TRATAMIENTO OPORTUNO | DEFICIENCIA ENZIMÁTICA | MIM o PS | FÓRMULA ESPECIAL | SUPLEMENTO, CONSIDERACIONES DE LA DIETA, Y OTROS TRATAMIENTOS | Referencias |
|---|---|---|---|---|---|---|
| AMINOÁCIDOS | ||||||
| Fenilcetonuria | Fenilalanina↑, discapacidad intelectual profunda e irreversible | Fenilalanina hidroxilasa | 261600 | 🡻fenilalanina y 🡹tirosina | L-tirosina, aminoácidos neutro largos, tetrahidropterina | 47, 48 |
| Hiperfenilalaninemia, deficiencia de BH4 | Fenilalanina↑.No responden adecuadamente a las fórmulas especiales con FA ↓, RDPM, DI, hipotonía axial e hipertonía apendicular, epilepsia | Dihidropteridina quinoide reductasa | 261630 | 🡻fenilalanina y 🡹tirosina | Tetrahidropterina: 2mg/kg | 48 |
| Tirosinemia Ia, Ib | Succinilacetona↑, tirosina↑, FA↑, metionina↑. Enfermedad hepática severa, trastorno tubular renal, raquitismo. | Fumarilacetoacetato | 276700 | 🡻fenilalanina y 🡹tirosina | NTBC 1-2 mg/kg/dia | 49 |
| Tirosinemia II | Tirosina ↑, FA normal. Úlceras corneales herpetiformes y queratosis punctata de dedos, palmas y plantas, DI. | Tirosina transaminasa | 276600 | 🡻fenilalanina y 🡹tirosina | Suplementación con ácido graso 3-omega | 50 |
| Enf. Orina con color a jarabe de arce | ↑Leucina, ↑isoleucina, ↑valina. Olor a jarabe de arce de cerilla, encefalopatía neonatal. | Transacilasa de cadena ramificada dihidrolipoamida, decarboxilasa BCKA subunidad beta, decarboxilasa BCKA subunidad alfa. | 248600 | 🡻Leucina | Tiamina oral: 100-300 mg/día. L-Valina, L-isoleucina | 50 |
| Acidemia isovalérica | Isovalerilacidemia, acidosis metabólica, RDPM, epilepsia, hemorragia cerebral, neutropenia, leucopenia, pancitopenia. | Isovaleril CoA dehidrogenasa | 243500 | 🡻Leucina | L-carnitina: 100 mg/kg/día. Glicina 200-400 mg/kg | 51 |
| Aciduria 3-OH-isobutírica | Acidemia orgánica, lactato↑, aciduria 3-OH-isobutiríca. | Defectos de la cadena respiratoria o defectos de la metilmalonato semialdehído dehidrogenasa | 236795 | 🡻valina | L-carnitina: 100 mg/kg/día | 52 |
| Aciduria 3-metilglutacónica | Aciduria 3 metilglutacónica. En la tipo 1 se observa falta de medro, atrofia óptica, cuadriplegia espástica, distonia, hiperreflexia, aciduria 3 metilglutacónica | 9 enzimas diferentes | PS250950 | 🡻Leucina | L-carnitina: 100 mg/Kg/día. Glicina 250-400 mg/Kg/día. Ácido pantonténico: 15-150 mg/día | 53 |
| Homocistinuria | Homocisteina↑, ectopia lentis, RDPM, DI | Cistationina β-sintasa | 236200 | 🡻metionina y 🡹cisteina | Ácido fólico: 500-1000/ mg/ 3 veces por día. Betaína: 150 mg/día. Piridoxina 25-750 mg/día. B12 1 mg(IM), 10-20 mg (oral) | 54 |
| Acidemia glutárica tipo 1 | Ácido glutárico↑, ácido glutacónico↑. Encefalopatía aguda, macrocefalia, lesiones de ganglios basales. | Glutaril CoA dehidrogenasa | 231670 | 🡻lisina y 🡻triptófano | Riboflavina: 100-300 mg/día | 55 |
| Intolerancia proteína lisinúrica | Lisina↑. Vómitos recurrentes, diarrea, episodios de coma, aversión a alimentos ricos en proteínas, hepatomegalia e hipotonía muscular | SLC7A7 (solute carrier familiy 7, member 7) | 222700 | 🡻ingesta de proteínas | L-citrulina: 2,5-8,5 g/día en 4 dosis | 56 |
| Acidemia propiónica y metilmalónica | Deterioro agudo, acidosis metabólica, amonio↑. Muerte temprana o trastorno neurológico, enfermedad crónica renal, cardiomiopatía | Metilmalonil CoA-mutasa y propionil CoA carboxilasa | 606054 y 251000 | 🡻metionina, isoleucina, treonina, valina | Biotina: 5-10 mg/día. B12 10-20 mg/día. L-Carnitina: 100 mg/kg/día. | 57 |
| Alteraciones del ciclo de la urea | Amonio↑↑, en casos con deficiencia enzimática severa se observa letargia, anorexia, hiper o hipoventilación, consvulsiones y coma. En casos con deficiencia leve, el amonio se eleva ocasionado por algún gatillo (enfermedad aguda o estrés), observándose pérdida del apetito, vómitos, letargia, delusiones, alucinaciones, psicosis y encefalopatía aguda | Carbamoil fosfato sintetasa I, ornitina transcarbamilasa, ácido arginino succínico sintetasa, ácido arginino succínico liasa, arginasa, N-acetil glutamato sintetasa, ornitina translocasa, citrina. | Heterogéneo | 🡻amomino | L-arginina: 200-400 mg/kg. L-citrulina: 200-400 mg/kg/día. Benzoato de sodio: 250-500 mg/kg/día, hemofiltración y hemodiálisis con ECMO, carbamil glutamato. | 58 |
| HIDRATOS DE CARBONO | ||||||
| Galactosemia clásica | Galactosa 1 fosfato↑. Problemas de deglución, falta de medro, daño hepatocelular, sangrado, sepsis por E. coli, RDPM, trastorno de lenguaje, falla ovárica prematura, catarata. | Galactosa 1-fosfato uridiltransferasa | 230400 | Eliminar galactosa (lactosa, galactolípidos) | Leche de soya. Calcio elemental | 59 |
| Glucogenosis tipo I | Hepatomegalia, nefromegalia, hipoglicemia, acidosis láctica, ácido úrico↑, lípidos↑, triglicéridos↑, convulsiones. Facies de "muñeca", talla baja, neutropenia crónica, xantoma, diarrea | Glucosa 6-fosfatasa o transportador de la glucosa 6-fosfato | 232200 | Eliminar lactosa, fructuosa, sorbitol | Hidratos de carbono compuestos: almidón crudo (1,5-2 g/kg/dosis). Fraccionamiento de la dieta | 60 |
| Intolerancia hereditaria a la fructuosa | Glucosa, acidemia láctica, fósforo ↓, ácido úrico ↑,, magnesio ↑, alanina↑. Naúseas, vómitos, falta de medro, letargia aguda, convusliones, coma. Falla hepática y renal. | Fructosa 1-fosfato aldolasa | 229600 | 🡻Fructuosa <10 mg/kg | Vitamina C | 61 |
| LÍPIDOS | ||||||
| Defectos de oxidación de ácidos grasos de cadena larga | Hipoglicemia, cetonas↓↓, insulina ↑, ácidos grasos libres↓. Cardiomiopatía, miopatia. | Transportador orgánico catiónico 2, carnitina palmitoil transferasa tipo 1A y 2, carnitina-acilcarnitina translocasa, VLCAD, proteína trifuncional mitocondrial | Heterogéneo | 🡻lípidos: 15-20% de las calorias totales | L-carnitina: 100 mg/kg. Fraccionamiento de la dieta. TCM: 30% del total de lípidos, DHA | 62 |
7. TRASPLANTE DE CÉLULAS HEMATOPOYÉTICAS
Conocido como como hematopoeitc stem cell transplatation (HSCT), el cual está ampliamente
utilizado en diferentes enfermedades genéticas. Este tipo de terapias está disponible y probado su
eficacia para inmunodeficiencias congénitas primarias (ej. enfermedad Duncan), osteogénesis imperfecta y
enfermedades de depósito lisosomal (LSD) (Figura 6), como la adrenoleucodistrofia
ligada al cromosoma X, mucopolisacaridosis I, II, VI y VII; leucodistrofia metacromática, fucosidosis y
manosidosis(64-67).
El fundamento para aplicar HSCT en las enfermedades de depósito lisosomal (LSD) se basa en la
capacidad que tienen las células trasplantadas y/o en su progenie celular (o clona) en contribuir a las
poblaciones de macrófagos de los tejidos afectados y así convertirse en fuentes permanentes locales de
enzimas lisosomales funcionales; de esta manera las células metabólicamente activas pueden mejorar el
fenotipo de la enfermedad al eliminar el material de almacenamiento y modular la inflamación local en
los sitios enfermos. El recambio de células con el donante después del trasplante se supone que afecta a
todos los tipos de poblaciones mieloides fijadas en tejido, incluidas las células mieloides y
posiblemente la microglía en el cerebro. Por esta razón, la HSCT fue pensada como una vía para tratar a
pacientes con deficiencias enzimáticas con afectación grave del sistema nervioso central (SNC). Es
importante destacar que, si se logra el quimerismo completo del donante, la HSCT es una intervención
única capaz de proporcionar una fuente de enzimas de por vida para el paciente afectado. Las células del
donante también restablecen un nuevo sistema inmunitario en el paciente, superando las preexistentes y
evitando las respuestas inmunitarias posteriores al tratamiento dirigidas a la enzima funcional. Sobre
esta base, desde que los primeros pacientes con LSD fueron trasplantados a principios de la década de
1980, unos pocos miles de pacientes con LSD han sido tratados con HSCT alogénico en las últimas
décadas(68). (Figura 6).
Es de suma importancia que la efectividad de la terapia dependerá en mayor o menor grado
mientras el paciente sea asintomático o mínimamente afectado(65,66).
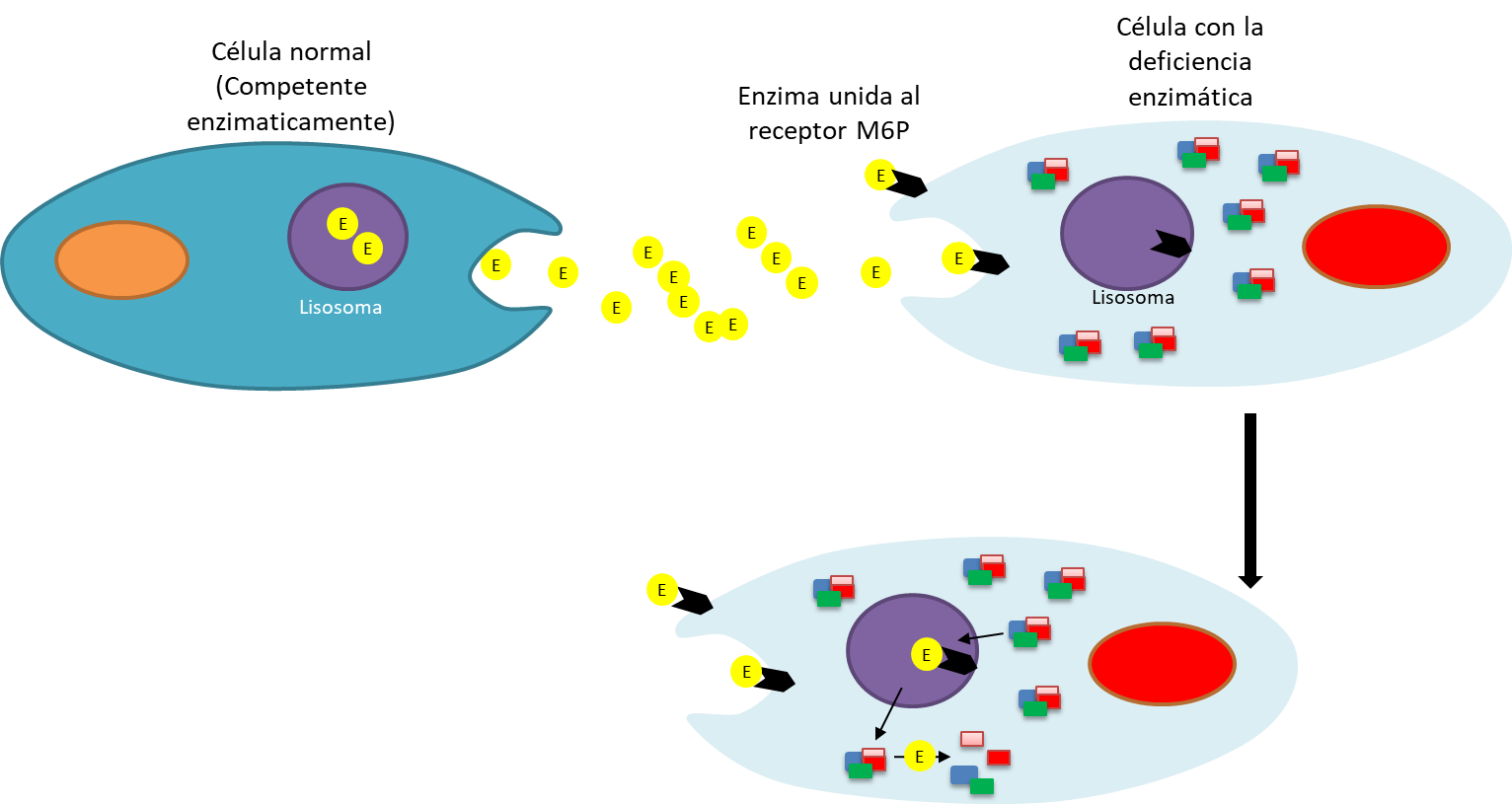
|
8. TERAPIA DE REEMPLAZO ENZIMÁTICO (TRE)
Existen muchas patologías de origen genético que entregando la proteína defectuosa cambiará la historia natural de la enfermedad. Dentro de este grupo de entidades se encuentran las enfermedades de depósito lisosomal y la deficiencia de adenosina desaminasa(69-78) (Tabla 2).
Tabla 2. Terapia de reemplazo enzimático en enfermedades genéticas.
| Entidad | MIM | Gen afectado | Características clínicas principales | TRE aprobado (FDA y/o EMA) | Referencias |
|---|---|---|---|---|---|
| Mucopolisacaridosis I | 607014, 607015, 607016 | IDUA | Facies tosca, macrocefalia, displasia esquelética, hepatoesplenomegalia, variable afectación neurológica, compromiso pulmonar y cardíaco, contracturas articulares progresivas, opacidad corneal. | Laronidasa | 74,75 |
| Mucopolisacaridosis II | 309900 | IDS | Facies tosca, macrocefalia, displasia esquelética, hepatoesplenomegalia, variable afectación neurológica, compromiso pulmonar y cardíaco, contracturas articulares progresivas, mano en garra. | Idursulfasa Hunterasa |
74,75 78 |
| Mucopolisacaridosis IV A | 253000 | GALNS | Facies tosca, displasia esquelética a predominio torácico, compromiso pulmonar y cardíaco, opacidad corneal, hiperlaxitud en manos. | Elosulfasa alfa | 74,75 |
| Mucopolisacaridosis VI | 253200 | ARSA | Facies tosca, macrocefalia, displasia esquelética, hepatoesplenomegalia, compromiso pulmonar y cardíaco, contracturas articulares progresivas, mano en garra. | Galsulfasa | 74,75 |
| Enfermedad Pompe | 232300 | GAA | Pérdida de fuerza muscular progresiva, al nacimiento se puede observar como hipotonia y cardiomegalia. | Alglucosidasa alfa | 70 |
| Enfermedad Gaucher | 230800 | GBA | Trombocitopenia, esplenomegalia, afectación ósea |
Imiglucerasa Velaglucerasa Taliglucerasa |
81 |
| Enfermedad Fabry | 301500 | AGA | Acroparestesias, angioqueratomas, enfermedad renal crónica, compromiso cardiaco, córnea verticilata, |
Agalsidasa alfa Agalsidada beta |
69,79 |
| Hipofosfatasia | 241500, 241510 | ALPL |
Disminución de la mineralización ósea y dental. Presentación variable desde fracturas patológicas, condrocalcinosis, “miopatía”, pérdida precoz de dientes deciduos, estatura corta Actividad disminuida de la fosfatasa alcalina ósea y sérica. |
Asfotasa alfa | 73 |
| Deficiencia de lipasa ácida lisosomal | 278000 | LIPA | Malnutrición, hepatomegalia con falla hepática, calcificación de las glándulas adrenales. Enfermedad de depósito de ésteres de colesterol. Cirrosis, hiperesplenismo, malabsorción intestinal. | Sebelipasa alfa | 76 |
| Deficiencia de adenosina desaminasa | 102700 | ADA | Inmunodeficiencia combinada severa por acumulación de metabolitos tóxicos que provocan un malfuncionamiento y formación de los linfocitos | PEG-ADA | 70 |
| Fenilcetonuria | 261600 | PAH | Utilizado en pacientes adultos con fenilcetonuria | Pegvaliasa | 77 |
| Lipofuscinosis neuronal ceroide tipo 2 | 204500 | TPP1 | Inicio a los 2-4 años de edad con epilepsia, neuroregresión, ataxia mioclónica, signos piramidales, discapacidad visual (4-6 años de edad). | Cerliponasa alfa |
9. CHAPERONAS
Actualmente aprobado el uso de migalastat en la enfermedad Fabry (MIM #301500). Las chaperonas tienen la función de estabilizar la actividad usual de una proteína(79).
10. TERAPIA DE REDUCCIÓN DEL SUSTRATO
La terapia de reducción de sustrato consiste en disminuir el o los metabolitos un paso antes de la vía afectada. El miglustat y el eliglustat se tiene como arma terapéutica para enfermedades como Gaucher 1 (MIM # 230800) y Niemann-Pick tipo C (MIM #257220) (80–82). Existen muchas revisiones que la genisteína tiene este mecanismo de acción en las mucopolisacaridosis (ej. tipo III)(83).
11. TERAPIA CROMOSÓMICA
Se encuentra en investigación básica y se basa en mejorar el efecto de las duplicaciones o
deleciones parciales o totales. Dentro de las estrategias utilizadas se tiene(84,85):
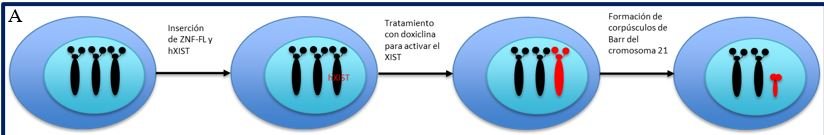
Silenciamiento de cromosomas con XIST, el cual consiste en utilizar nucleasas (ej. ZNF) para
insertar una forma inducible del gen XIST en una de las copias en las células trisómicas (Figura 7A).
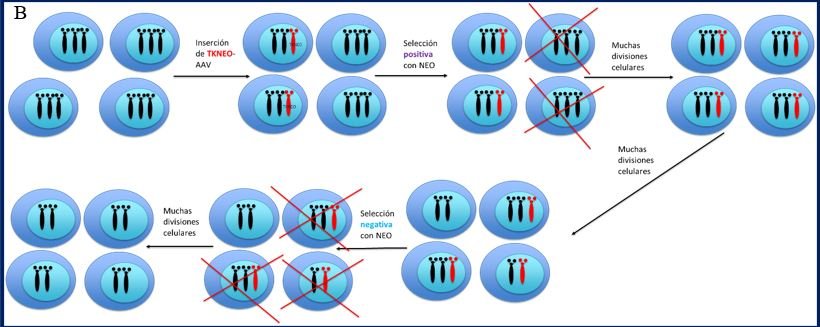
11a.- Marcadores seleccionables positivo-negativo en el cromosoma extra; se usa el transgén de
timidina quinasa-neomicina (TKNEO), quien ayuda a seleccionar con antibióticos y luego aislar células
disómicas de una población trisómica. Las células totalmente trisómicas (iPSCs-células madre
pluripotentes inducibles) son infectadas con un vector viral adeno-asociado (AAV) que contiene un
transgén TKNEO el que confiere resistencia a la neomicina (NEO) y sensibilidad al ganciclovir. Debido a
la eficiencia imperfecta, sólo algunas células de la población reciben el transgen TKNEO. La población
celular se trata con neomicina, posterior a esto se eliminan las células que no contienen el transgen
TKNEO. La población que contiene el transgén puro se prolifera para permitir que ocurran eventos de no
disyunción de manera natural. Luego la cohorte de células disómicas y trisómicas se trata con
ganciclovir (GCV); eliminándose todas las células trisómicas y que contienen el transgen TKNEO dejando
sólo la población disómica pura que puede ser aislada y proliferada (Figura 7B).
11b.- Rescate trisómico inducido por drogas; donde se cultiva células trisómicas (trisomía 21 y
18) con ZSCAN4, el cual incrementa el número de células euploides (normales) en un 24%.
11c.- Cromosomas humanos artificiales (HAC-human artificial chromosomes); conocidos como
minicromosomas, los que se utilizan como “vectores". Estos se integran libremente al ciclo celular en el
tiempo, el cual tendría la posibilidad de corregir las deleciones.
11d.- Inducción de formación de cromosomas en anillo. A las células trisómicas (iPSCs) se
inserta LoxP en el brazo corto y largo del cromosoma a través de CRISPR-Cas9. Luego las células con
tratadas con una recombinasa que induce la formación de cromosomas en anillo, luego estas células se
replican y pierden al cromosoma en anillo, de forma natural, restableciendo el estado disómico.
e.- Inhibición del gen DYRK1A, se ha demostrado que este gen se ve implicado en la
fisiopatología de la discapacidad intelectual del síndrome Down. Una de las drogas que se demostró su
eficacia y seguridad en pacientes adultos (fase 2) con síndrome Down es el de la
epigalatocatequina-3-galato (extracto del té verde), mejorando la cognición, memoria de reconocimiento
visual, control inhibitorio y del comportamiento adaptativo(86,87).
 |
 |
12. OTRAS TERAPIAS
Algunas enfermedades monogénicas en la actualidad cuentan con terapias en investigación clínica, lo cual
podría ser verificado en el portal www.clinicaltrials.com.
Algunas de las terapias mostradas probablemente no estén enfocadas directamente a lo descrito en
la figura 1; sin embargo, se ha visto que tienen una utilidad enorme en el manejo de
estas enfermedades.
Distrofia muscular Duchenne (DMD), es una patología que se manifiesta por pérdida progresiva de
fuerza muscular en la primera década. Actualmente se tiene disponible tres terapias, una de ellas la
mencionamos en terapias por nucleótidos cortos, y las otras es el uso de deflazacort y el ataluren. El
deflazacort está usado ampliamente en la DMD desde hace más de 30 años; sin embargo, su aprobación por
FDA fue recién desde el año 2017(88).
El ataluren se utiliza en aquellos pacientes que tienen una variante sin sentido (10-15% de los
pacientes con DMD). Su mecanismo de acción es realizar un salto de lectura en el lugar de la variante
sin sentido, haciendo que la proteína sea de mayor tamaño a la proteína “mutada”. De esta manera lo que
provoca es cambiar el fenotipo a la distrofia muscular Becker(89).
En este mismo sentido, el uso de bifosfonatos en enfermedades como osteogénesis imperfecta
(PS166200) y síndrome McCune-Albright (MIM #174800) están indicados para disminuir el dolor y el riesgo
de aparición de fracturas(90,91).
Otras terapias en la osteogénesis imperfecta que se ha observado que disminuye el riesgo de
fracturas es a través de la activación de osteoclastos (denosumab), agentes anabólicos óseos
(teriparatide, romosozumab)(67).
En el raquitismo hipofosfatémico ligado al cromosoma X (MIM #307800) es una condición donde se
observa una hipofosfatemia crónica la que ocasiona una falla en la mineralización provocando raquitismo
y osteomalacia. Se ha observado que un inhibidor monoclonal del FGF23 (burosumab) es una terapia
prometedora en esta condición(92).
La esclerosis tuberosa (MIM #PS191100) tiene manifestaciones clínicas muy heterogéneas y la FDA
ha aprobado el uso de inhibidores mTOR como el everolimus para la epilepsia, rabdomiosarcomas,
astrocitomas, angiomiolipomas; y la rapamicina para linfangioleiomatosis(93).
Conclusiones
La farmacopea en las enfermedades genéticas se incrementa notablemente en el tiempo. Muchas terapias
tratan de ser muy específicas; sin embargo, se están desarrollando medicinas que se utilizarán en más de
una entidad, que incluso no tienen relación etiológica.
Como se está viendo en estos últimos años, estas nuevas terapias están cambiando la historia
natural de este grupo de entidades. No obstante, el cuello de botella en estas condiciones es el
diagnóstico, ya sea por el número limitado de especialistas, falta de implementación, costos elevados,
coberturas por parte de las aseguradoras, entre otras.
El futuro de la medicina en general está condicionado a entender de mejor manera los mecanismos
subyacentes e inherentes de cada enfermedad, basada en una comprensión individual de nuestra “ómica”,
llevándolo por lo tanto a otro nivel de medicina: Medicina de precisión.
Finalmente, es importante indicar que todos estas terapias y medicamentos, son opciones
terapéuticas prometedoras y valiosas para estas diferentes enfermedades descritas; no obstante, es de
suma importancia que el manejo de todas estas condiciones es multi e interdisciplinario y llevadas a
cabo por profesionales calificados dentro de laboratorios e instituciones adecuadamente certificadas
para estos propósitos.
Contribuciones de Autoría: Todos los autores participaron en el diseño del estudio,
recolección de información, y en la redacción del manuscrito.
Financiamiento: Ninguno
Conflictos de intereses: Ninguno declarado por los autores.
Recibido: 07 de abril 2020
Aprobado: 01 de diciembre 2020
Correspondencia: Hugo Hernán Abarca Barriga.
Dirección: Servicio de Genética & EIM, Instituto Nacional de Salud del Niño, Av.
Brasil 600, CP Lima 05, Lima, Perú.
Teléfono: +51 979301132
Correo: habarca@insn.gob.pe
REFERENCIAS BIBLIOGRÁFICAS
