ARTICULO REVISIÓN
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2021 - Universidad Ricardo Palma10.25176/RFMH.v21i2.3626
MANAGEMENT OF GENETIC DISEASES: PRESENT AND FUTURE
TRATAMIENTO DE LAS ENFERMEDADES GENÉTICAS: PRESENTE Y FUTURO
Hugo Hernán Abarca Barriga(1,2,3,a,b), Milana Trubnykova(2,a), María del Carmen Castro Mujica (1,a)
1 Facultad de Medicina Humana, Universidad Ricardo Palma, Lima-Perú.
2 Servicio de Genética & EIM, Instituto Nacional de Salud del Niño-Breña, Lima-Perú.
3 Universidad Científica del Sur, Lima-Perú
a MD Specialist in Medical Genetics
b Master in Genetics
ABSTRACT
Today, the number of genetic diseases is around 10000 conditions, affecting to 6%-8% of all populations. This review shows us how the discovery of genetic variants in our genome, this facilitated to know with precision about the mechanisms physiopathological, and hence to recognize those target points susceptible to modifications, through therapeutical strategies different with palliative proposals, increase life expectancy, or improve qualities of life. These therapies are diverse, using drugs for polygenic diseases, nutritional therapy, special formulas, enzyme replacement therapies, hematopoietic stem cell transplant, substrate reduction, oligonucleotides, and gene therapy. These genetic diseases are heterogeneous clinically with a very low frequency; nevertheless, open to the possibility of research in new strategies for more genetic disease, that today, furthermore, are orphans.
Keywords: Genetic diseases; genetic Therapy, hematopoietic Stem Cells, transplant, therapy (Source: MeSH NLM)
RESUMEN
El número de enfermedades genéticas se estima que podrían ser más de 10 000 condiciones diferentes, afectando alrededor del 6-8% de la población. La presente revisión nos muestra la importancia del descubrimiento de las variantes patogénicas en nuestro genoma que nos permite conocer con mayor precisión cuales son los mecanismos fisiopatológicos y, por lo tanto, conocer puntos dianas susceptibles de modificaciones mediante diferentes estrategias terapéuticas para poder palear los síntomas y signos, aumentar la expectativa de vida, mejorando así la calidad de vida de los pacientes que tienen algunas de estas enfermedades genéticas. Las diferentes terapias que existen en la actualidad son muy diversas como fármacos de uso en patologías comunes, terapia nutricional, fórmulas especiales, terapias de reemplazo enzimático, trasplante de órganos y células hematopoyéticas, reducción de sustrato, oligonucleótidos y la terapia génica. Al ser las enfermedades genéticas clínicamente heterogéneas, abre la posibilidad de poder investigar cada vez más nuevas estrategias en un mayor número de enfermedades que en la actualidad están olvidadas.
Palabras Clave: Enfermedades genética, terapia genética, Células Madre hematopoyéticas, trasplantes, terapias (Fuente: DeCS BIREME)
INTRODUCTION
A rare disease is defined by the appearance frequency. That is why, for example, in Europe it is
referred as the one with an incidence of less than 1/2000 people. The number of patients who are
affected is estimated between 6% to 8% of the general population. In our country there are no studies
that define the real number of affected people, therefore it is estimated that they are approximately 2
million of Peruvian people((1)). Nonetheless, some studies that were done
estimate that the percentage of affected people by a rare disease may be between 3.5% to 5.9%.
The etiology of rare disease has a genetic origin in 80% of the total cases, and the other 20%
has an unknown origin. The genetic origin can be divided into three groups: i) the ones that are
produced by variants in a unique nucleotide (SNV, nucleotide variant), ii) variants of multiple
nucleotides (MNV, multinucleotide variant) and iii) variants in the number of copies (CNV, copy number
variation). The first two variants mainly produce monogenic diseases, which are estimated by the World
Health Organization (WHO) to number more than 10,000 entities((1)). Pathogenic
(or probably pathogenic) CNVs that cause microdeletion/microduplication syndromes; where the most
frequent have a prevalence between 1/1 000 to 1/25 000((3)); although, it has
been reported that in fetuses the incidence of CNVs is higher reaching 0.7%((4)). It is important to clarify that not all genetic diseases are rare. (e.g.
Down syndrome, Klinefelter syndrome)((5)). Of this large group of conditions,
about 500 diseases have a targeted treatment((6)).
It should be noted that genetic diseases account for up to 71% of pediatric
hospitalizations((7)) and cause between 20% and 30% of deaths in this age
group((8)). This proportion of patients generates a large economic impact on
health systems; thus, an Australian study carried out in a population-based cohort in 2010 found that
patients with rare diseases generated 10.5% of hospital expenses((9)) , in
addition to a longer hospital stay than their peers without genetic conditions((7)).
The clinical manifestations of genetic diseases are very diverse, i.e. they have a great
clinical or phenotypic variability and can manifest as hypotonia, delayed psychomotor development,
intellectual disability, epilepsy, neuroregression, congenital anomalies, short stature, microcephaly,
primary immunodeficiencies, schizophrenia, autism spectrum disorders, conduct disorders, attention
deficit hyperactivity disorder, dementia, abnormal movements and cancer. There are even entities, such
as infantile cerebral palsy, in which a genetic component was not previously described and it is now
considered that up to 20% of cases have a genetic cause(1). It is important to
point out that genetic diseases can appear at any stage of life, from prenatal to adulthood(10).
Since the end of the 20th century, thanks to the decoding of DNA and a better understanding of
the pathophysiology of genetic diseases, targeted therapies, namely those that are directed at the
factor or factors that initiate the disease, have been progressively and steadily increasing, which is
aided by bioinformatics(11).
Therapies for genetic diseases are available and their use has been approved by international
institutions such as the Food and Drug Administration (FDA)(12,13) and the European Medicine Agency (EMA)(14,15). On the other hand, there is a great expectation of new treatments which
are in basic research and some of them in clinical research, as can be seen in the clinical trials
portal with more than 2,520 different studies(16).
In this review we aim to try to identify in a general way the pharmacological treatment
currently existing and what is being investigated in these genetic diseases. The way in which a
therapeutic approach is carried out is focused on one of the points of the pathophysiological cascade of
genetic diseases. Thus, treatment could be at the level of the affected gene(s) (e.g. gene and
chromosome therapy), replacing the abnormal protein (e.g. hematopoietic cell transplantation), modifying
the metabolic cascade (e.g. special formulations, substrate reduction therapy) and symptomatic(17) (Figure 1)

|
1. GENE THERAPY
The main objective of gene therapy (also known as gene therapy) is to sufficiently incorporate a
long-lasting expression of a therapeutic gene or transgene in order to improve or cure symptoms with
minimal adverse events(18).
When research began, it focused mainly on monogenic diseases. However, currently most clinical
research studies are directed at cancer(19) (Figure 2).
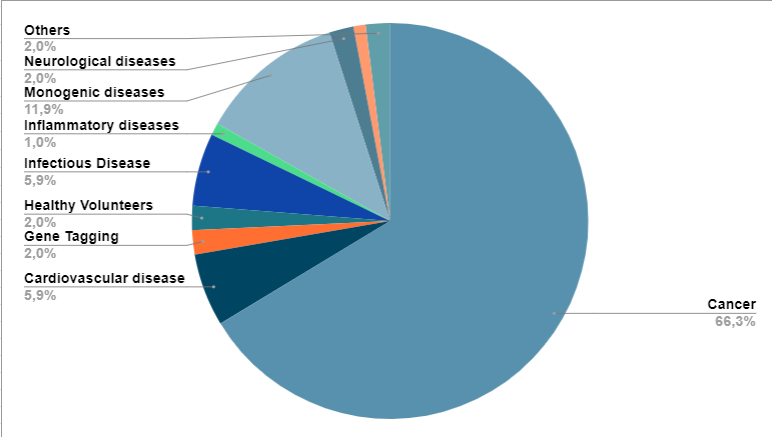
|
The types of gene therapies are directed to germ cells (sperm or egg cells) or somatic cells. The
duration of the expression of the transferred gene depends on the type of pathology, for example, in
monogenic diseases the time should be prolonged, while in multifactorial diseases (e.g. cancer,
infectious diseases) it should be short(20).
There are two types of gene transfer: in-vivo and ex-vivo(21). The
former means that the gene is delivered directly to a tissue, while in the latter, cells are extracted
from the patient, the gene is delivered and then incorporated back into the affected individual(18,21). The types of gene therapy can be subdivided into
those using virus-mediated therapy and nanoparticles, synthetic short nucleotides, as well as gene
editing(22).
2. VIRUS-BASED THERAPIES.
The most frequently used viruses are: adenovirus, adeno-associated viruses, lentivirus, retrovirus(23,24) (Figure 3). Adeno-associated viruses are the most commonly used because they have a greater capacity to infect different tissues and have a lower inflammatory response(20).
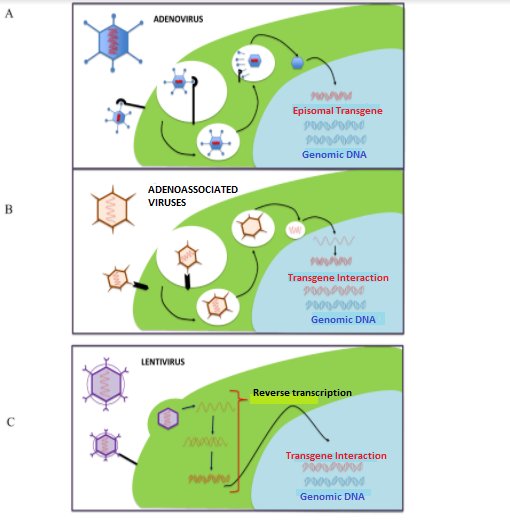
|
Since 2016 to date, virus-based genotherapies have been approved (by FDA and EMA), which we mention below(18,25):
- Alipogene tiparvovec -Glybera- is an adeno-associated virus (AAV1) used for hyperlipoproteinemia type 1 (MIM #238600) caused by recessive variants of the LPL gene, leading to lipoprotein lipase deficiency, causing hyperchylomicronemia and pancreatitis(26).
- Strimvelis, uses a retrovirus as a vector, which is used in adenosine deaminase deficiency (ADA gene), characterized by severe combined immunodeficiency (MIM #102700)(27).
- Zynteglo, using a lentivirus as a vector, is used in beta-thalassemia (MIM #613985), characterized by congenital hypochromic microcytic anemia, decreased hemoglobin (Hb) A and increased Hb F, hepatosplenomegaly(28).
- Voretigene neparvovec-rzyl -Luxturna- (AAV2), approved for the use of recessive variants of the RPE65 gene that causes Leber congenital amaurosis (MIM #204100) and retinitis pigmentosa 20 (MIM #613794)(29).
- Onasemnogene abeparvovec-xioi -Zolgensma- (AAV9), which is used for spinal muscular atrophy 1 (MIM #253300), who are children presenting progressive congenital hypotonia, where the majority of those affected (95-98%) present a deletion in exon 7 of the SMN1 gene(30).
3. THERAPIES WITH SHORT NUCLEOTIDES
Among the therapies that use short synthetic nucleotides, there are two types:
- Antisense oligonucleotides (AON, from antisense oligonucleotide), have 20-30 nucleotides of DNA, with two forms of action: i) using RNA Hase, in which it destroys messenger RNA (mRNA) and ii) without using RNA Hase, where it can act by modulating splicing, through steric blocking, binding to the 5' cap region of mRNA or the 3' poly A region(22,31-33) (Figure 4A).
- ARN de interferencia (ARNi), se utilizan como mecanismo de defensa natural contra los virus ARN. El mecanismo de acción es mediante la utilización de los complejos moleculares Dicer (ribonucleasa) y RISC (del inglés, RNA-induced silencing complex), uniéndose de manera complementaria al ARNm y su posterior rompimiento(22,31-33) (Figure 4B).
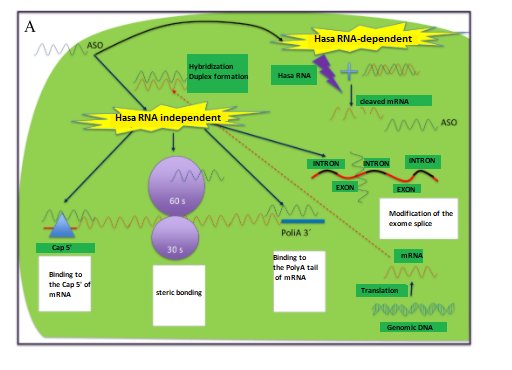 |
 |
To date, we have the following molecules approved by the FDA and/or EMA:
- Eteplirsen: is an AON used in patients with Duchenne muscular dystrophy (MIM #310200) and who present the deletion of exon 51 of the DMD gene; causing a skipping of this exon resulting in a short protein, however, with greater functionality(34,35).
- Nusinersen: is an AON used in spinal muscular atrophy type 1 (MIM #253300), which is caused by homozygous variants of the SMN1 gene. This AON is used in patients who have at least one copy of the SMN2 gene, modifying the expression of the SMN2 gene (which is usually decreased), being a protein similar to SMN1(36-38).
- Paitisiran: is an RNAi used in transthyretin-related hereditary amyloidosis (MIM #105210), caused by heterozygous monoallelic variants in the TTR gene. This RNAi causes the reduction of the "mutant" protein(39).
- Mipomersen: is an AON used in familial hypercholesterolemia (variants in the LDLR, APOB, PCSK9 genes)(40).
4. GENETIC/GENE EDITION
On the other hand, it is of utmost importance to know that greater possibilities are opening up with the use of gene editing through meganucleases, nucleases such as ZNF (zinger nuclear finger), TALE (transcription activator-like repeat) and CRISPR/Cas9 (clustered regularly interspaced short palindromic repeat / CRISPR associated protein 9). The latter system is based on a system found in bacteria and archae, which confers resistance to viruses. CRISPR/Cas 9 contains two elements, an endonuclease (Cas 9) and a simple guide sequence (sgRNA) (Figure 5A). Uses range from gene regulation (Figure 5B-5E), epigenetic modification to genome imaging. Monogenic diseases under basic research include congenital cataract, Duchenne muscular dystrophy, hereditary tyrosinemia type 1, cystic fibrosis, betatalasemia, urea cycle disorders(41).
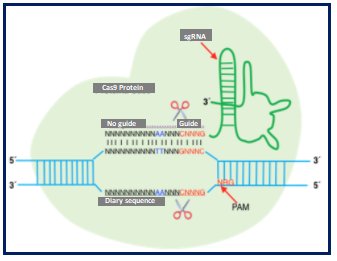 |
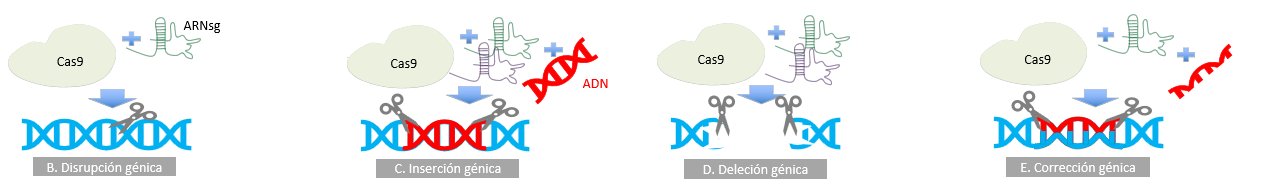 |
5. GENE THERAPY USING NON-VIRAL VECTORS
These are research strategies that have the possibility of incorporating DNA through synthetic vectors, which are frequently known as nanoparticles (NPs) measuring 10 to 500 nm(23). These NPs have the advantage of very easy synthesis, lower production costs than viral vectors, greater safety, the capacity to transport larger molecules and greater efficacy. (23) These nanoparticles can be composed of polysaccharides, solid lipids or coated with CK30PEG (30-mer cationic polylysine conjugated with 10KDa polyethilene glycol)(23.) On the other hand, the incorporation of DNA is being tested with the use of a vector ("naked" DNA) by means of physical methods such as electroporation, sonoporation, magnetofection and "bullet" genes(20) .
6. NUTRITIONAL THERAPY
This type of therapy is mainly used for inborn errors of metabolism (IEM)(42). It is important to emphasize that there are at least 81 pathologies
that, with early diagnosis and timely treatment, will prevent the risk of intellectual disability
(www.treatable-id.org)(43). It is of utmost importance to emphasize that the
ideal moment of diagnosis is as early as possible, and if possible through universal neonatal screening
of at least the most frequent entities(44). We can divide this type of
therapy into(45,46) (Table 1):
6a.- Nutrient restriction
When it is known that there is an increase in a toxic metabolite due to a decrease in enzymatic
activity, and that there are other metabolites cascading above; what is done is to reduce these through
special formulas, causing the toxic to decrease, thus avoiding the onset of the pathophysiological
cascade(47,48).
6b.- Nutritional supplementation
In many cases, apart from nutrient restriction with special formulas, it is necessary to supplement
with metabolites that are not adequately produced(45,46).
6c.- Elimination or blocking of toxic metabolite synthesis
There are many IEM, where the pathophysiology of the picture is mainly framed in the alternative
production of a toxic metabolism, so it is necessary to use drugs or procedures (e.g. the use of
hemofiltration in urea cycle defects) that eliminate or block the synthesis of these(45,46).
Table 1. Management of some inborn errors of amino acid, carbohydrate and lipid metabolism.
| DISEASE | CLINICAL FEATURES WITHOUT TIMELY TREATMENT | ENZYME DEFICIENCY | MIM o PS | SPECIAL FORMULATION | SUPPLEMENTATION, DIETARY, CONSIDERATIONS AND OTHER TREATMENTS | REFERENCES |
|---|---|---|---|---|---|---|
| AMINO ACIDS | ||||||
| Phenylketonuria | Phenylalanine↑, profound and irreversible intellectual disability. | Phenylalanine hydroxylase | 261600 | 🡻phenylalanine and 🡹tyrosine | L-tyrosine, long neutral amino acids, tetrahydropterin | 47, 48 |
| Hyperphenylalaninemia, BH4 deficiency | Phenylalanine↑.Do not respond adequately to special formulations with FA ↓, RDPM, DI, axial hypotonia and appendicular hypertonia, epilepsy. | Dihydropteridine quinoid reductase | 261630 | 🡻phenylalanine and 🡹tyrosine | Tetrahydropterin: 2mg/kg | 48 |
| Tyrosinemia Ia, Ib | Succinylacetone↑, tyrosine↑, FA↑, methionine↑. Severe liver disease, renal tubular disorder, rickets. | Fumarylacetoacetate | 276700 | 🡻phenylalanine and 🡹tyrosine | NTBC 1-2 mg/kg/day | 49 |
| Tyrosinemia II | Tyrosine ↑, normal FA. Herpetiform corneal ulcers and punctate keratosis of fingers, palms and soles, DI. | Tyrosine transaminase | 276600 | 🡻phenylalanine and 🡹tyrosine | 3-omega fatty acid supplementation | 50 |
| Maple syrup-colored urine disease | ↑Leucine, ↑isoleucine, ↑valine. Matchstick maple syrup odor, neonatal encephalopathy. | Dihydrolipoamide branched-chain transacylase, BCKA beta-subunit decarboxylase, BCKA alpha-subunit decarboxylase. | 248600 | 🡻Leucine | Oral thiamine: 100-300 mg/day. L-Valine, L-isoleucine | 50 |
| Isovaleric acidemia | Isovalerylacidemia, metabolic acidosis, RDPM, epilepsy, cerebral hemorrhage, neutropenia, leukopenia, pancytopenia. | Isovaleryl CoA dehydrogenase | 243500 | 🡻Leucine | L-carnitine: 100 mg/kg/day. Glycine 200-400 mg/kg | 51 |
| 3-OH-isobutyric aciduria | Organic acidemia, lactate↑, 3-OH-isobutyric aciduria. | Defects of the respiratory chain or defects of methylmalonate semialdehyde dehydrogenase. | 236795 | 🡻valine | L-carnitine: 100 mg/kg/day. | 52 |
| 3-Methylglutaconic aciduria | 3-methylglutaconic aciduria. In type 1, lack of medro, optic atrophy, spastic quadriplegia, dystonia, hyperreflexia, 3-methylglutaconic aciduria are observed. | 9 different enzymes | PS250950 | 🡻Leucine | L-carnitine: 100 mg/kg/day. Glycine 250-400 mg/kg/day. Pantontenic acid: 15-150 mg/day. | 53 |
| Homocystinuria | Homocysteine↑, ectopia lentis, RDPM, DI | Cystathionine β-synthase | 236200 | 🡻methionine and 🡹cysteine | Folic acid: 500-1000/ mg/ 3 times per day. Betaine: 150 mg/day. Pyridoxine 25-750 mg/day. B12 1 mg (IM), 10-20 mg (oral). | 54 |
| Glutaric acidemia type 1 | Glutaric acid↑, glutaconic acid↑. Acute encephalopathy, macrocephaly, basal ganglia lesions. | Glutaryl CoA dehydrogenase | 231670 | 🡻lysine and 🡻tryptophan | Riboflavin: 100-300 mg/day. | 55 |
| Lysinuric protein intolerance | Lysine↑. Recurrent vomiting, diarrhea, coma episodes, aversion to protein-rich foods, hepatomegaly and muscular hypotonia. | SLC7A7 (solute carrier familiy 7, member 7) | 222700 | 🡻protein intake | L-citrulline: 2.5-8.5 g/day in 4 doses. | 56 |
| Propionic and methylmalonic acidemia | Acute deterioration, metabolic acidosis, ammonium↑. Early death or neurologic disorder, chronic renal disease, cardiomyopathy. | Methylmalonyl CoA-mutase and propionyl CoA carboxylase | 606054 y 251000 | 🡻methionine, isoleucine, threonine, valine | Biotin: 5-10 mg/day. B12 10-20 mg/day. L-Carnitine: 100 mg/kg/day. | 57 |
| Urea cycle disorders | Ammonium↑↑↑, in cases with severe enzyme deficiency, lethargy, anorexia, hyper- or hypoventilation, consvulsions and coma are observed. In cases with mild deficiency, ammonium is elevated due to a trigger (acute illness or stress), with loss of appetite, vomiting, lethargy, delusions, hallucinations, psychosis and acute encephalopathy. | Carbamoyl phosphate synthetase I, ornithine transcarbamylase, argininosuccinic acid synthetase, argininosuccinic acid lyase, arginase, N-acetyl glutamate synthetase, ornithine translocase, citrinase. | Heterogeneous | 🡻amino | L-arginine: 200-400 mg/kg. L-citrulline: 200-400 mg/kg/day. Sodium benzoate: 250-500 mg/kg/day, hemofiltration and hemodialysis with ECMO, carbamyl glutamate. | 58 |
| CARBOHYDRATES | ||||||
| Classic galactosemia | Galactose 1-phosphate↑. Swallowing problems, failure to thrive, hepatocellular damage, bleeding, E. coli sepsis, RDPM, language disorder, premature ovarian failure, cataract. | Galactose 1-phosphate uridylyltransferase | 230400 | Eliminate galactose (lactose, galactolipids) | Soy milk. Elemental calcium | 59 |
| Glycogenosis type I | Hepatomegaly, nephromegaly, hypoglycemia, lactic acidosis, uric acid↑, lipids↑, triglycerides↑, seizures. "Doll" facies, short stature, chronic neutropenia, xanthoma, diarrhea. | Glucose 6-phosphatase or glucose 6-phosphate transporter | 232200 | Eliminate lactose, fructose, sorbitol | Compound carbohydrates: raw starch (1.5-2 g/kg/dose). Dietary fractionation | 60 |
| Hereditary fructose intolerance | Glucose, lactic acidemia, phosphorus ↓, uric acid ↑,, magnesium ↑, alanine↑. Nausea, vomiting, lack of medro, acute lethargy, convulsions, coma. Hepatic and renal failure. | Fructose 1-phosphate aldolase | 229600 | 🡻Fructose <10 mg/kg | Vitamin C | 61 |
| LIPIDS | ||||||
| Long-chain fatty acid oxidation defects | Hypoglycemia, ketones↓↓↓, insulin ↑ free fatty acids↓. Cardiomyopathy, myopathy. | Cationic organic transporter 2, carnitine palmitoyltransferase type 1A and 2, carnitine-acylcarnitine translocase, VLCAD, mitochondrial trifunctional protein | Heterogeneous | 🡻Lipids: 15-20% of total calories | L-carnitine: 100 mg/kg. Dietary fractionation. MCT: 30% of total lipids, DHA | 62 |
7. HEMATOPOIETIC CELL TRANSPLANTATION
Known as hematopoietic stem cell transplantation (HSCT), which is widely used in different genetic
diseases. This type of therapy is available and proven effective for primary congenital
immunodeficiencies (e.g. Duncan disease), osteogenesis imperfecta and lysosomal storage diseases (LSD)
(Figure 6), such as X-linked adrenoleukodystrophy, mucopolysaccharidosis I, II, VI
and VII; metachromatic leukodystrophy, fucosidosis and mannosidosis(64-67).
The rationale for applying HSCT in lysosomal storage diseases (LSD) is based on the ability of
transplanted cells and/or their cell progeny (or clones) to contribute to the macrophage populations of
affected tissues and thus become permanent local sources of functional lysosomal enzymes; in this way
metabolically active cells can improve the disease phenotype by removing storage material and modulating
local inflammation at diseased sites. Cell turnover with the donor after transplantation is supposed to
affect all types of tissue-bound myeloid populations, including myeloid cells and possibly microglia in
the brain. For this reason, HSCT was intended as an avenue for treating enzyme-deficient patients with
severe central nervous system (CNS) involvement. Importantly, if complete donor chimerism is achieved,
HSCT is a unique intervention capable of providing a lifelong source of enzymes for the affected
patient. The donor cells also re-establish a new immune system in the patient, overcoming pre-existing
ones and preventing post-treatment immune responses directed at the functional enzyme. On this basis,
since the first LSD patients were transplanted in the early 1980s, a few thousand LSD patients have been
treated with allogeneic HSCT over the past decades(68). (Figure 6).
It is of utmost importance that the effectiveness of therapy will depend to a greater or lesser
degree as long as the patient is asymptomatic or minimally affected(65,66).
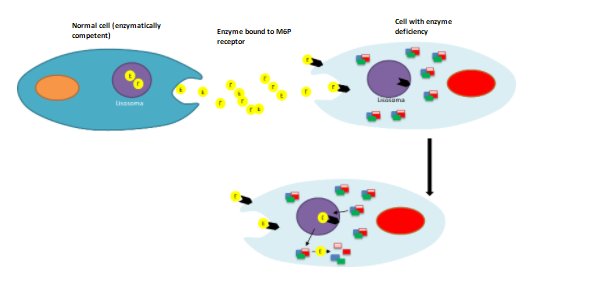
|
8. ENZYME REPLACEMENT THERAPY (ERT)
There are many pathologies of genetic origin that by delivering the defective protein will change the natural history of the disease. Within this group of entities are lysosomal depot diseases and adenosine deaminase deficiency(69-78) (Table 2).
Table 2. Enzyme replacement therapy in genetic diseases.
| Entity | MIM | Affected gene | Main clinical characteristics | Approved TRE (FDA and/or EMA) | References |
|---|---|---|---|---|---|
| Mucopolysaccharidosis I | 607014, 607015, 607016 | IDUA | Coarse facies, macrocephaly, skeletal dysplasia, hepatosplenomegaly, variable neurological involvement, pulmonary and cardiac involvement, progressive joint contractures, corneal opacity. | Laronidase | 74,75 |
| Mucopolysaccharidosis II | 309900 | IDS | Coarse facies, macrocephaly, skeletal dysplasia, hepatosplenomegaly, variable neurological involvement, pulmonary and cardiac involvement, progressive joint contractures, claw hand. | Idursulfase | 74,75 |
| Hunterase | 78 | ||||
| Mucopolysaccharidosis IV A | 253000 | GALNS | Coarse facies, skeletal dysplasia predominantly thoracic, pulmonary and cardiac involvement, corneal opacity, hyperlaxity in hands. | Elosulfase alfa | 74,75 |
| Mucopolysaccharidosis VI | 253200 | ARSA | Coarse facies, macrocephaly, skeletal dysplasia, hepatosplenomegaly, pulmonary and cardiac involvement, progressive joint contractures, claw hand. | Galsulfase | 74,75 |
| Pompe Disease | 232300 | GAA | Progressive loss of muscle strength, at birth can be observed as hypotonia and cardiomegaly. | Alglucosidase alfa | 70 |
| Gaucher Disease | 230800 | GBA | Thrombocytopenia, splenomegaly, bone involvement. | Imiglucerase | 81 |
| Velaglucerase | |||||
| Taliglucerase | |||||
| Fabry Disease | 301500 | AGA | Acroparesthesias, angiokeratomas, chronic renal disease, cardiac involvement, cornea verticilata, | Agalsidase alpha | 69,79 |
| Agalsidase beta | |||||
| Hypophosphatasia | 241500, 241510 | ALPL | Decreased bone and dental mineralization. Variable presentation from pathological fractures, chondrocalcinosis, "myopathy", early loss of deciduous teeth, short stature. | Asfotase alpha | 73 |
| Decreased bone and serum alkaline phosphatase activity. | |||||
| Lysosomal acid lipase deficiency | 278000 | LIPA | Malnutrition, hepatomegaly with hepatic failure, calcification of the adrenal glands. Cholesterol ester deposition disease. Cirrhosis, hypersplenism, intestinal malabsorption. | Sebelipase alfa | 76 |
| Adenosine deaminase deficiency | 102700 | ADA | Severe combined immunodeficiency due to accumulation of toxic metabolites causing malfunction and formation of lymphocytes. | PEG-ADA | 70 |
| Phenylketonuria | 261600 | PAH | Used in adult patients with phenylketonuria. | Pegvalase | 77 |
| Neuronal ceroid lipofuscinosis type 2 | 204500 | TPP1 | Onset at 2-4 years of age with epilepsy, neuroregression, myoclonic ataxia, pyramidal signs, visual impairment (4-6 years of age). | Cerliponase alpha |
9. CHAPERONAS
Migalastat is currently approved for use in Fabry disease (MIM #301500). Chaperones have the function of stabilizing the usual activity of a protein(79).
10. TERAPIA DE REDUCCIÓN DEL SUSTRATO
Substrate reduction therapy consists of reducing the metabolite(s) one step upstream of the affected pathway. Miglustat and eliglustat are held as therapeutic weapons for diseases such as Gaucher 1 (MIM # 230800) and Niemann-Pick type C (MIM #257220) (80–82). There are many reviews that genistein has this mechanism of action in mucopolysaccharidoses (e.g. type III)(83).
11. CHROMOSOME THERAPY
Se encuentra en investigación básica y se basa en mejorar el efecto de las duplicaciones o deleciones
parciales o totales. Dentro de las estrategias utilizadas se tiene(84,85):
Silenciamiento de cromosomas con XIST, el cual consiste en utilizar nucleasas (ej. ZNF) para
insertar una forma inducible del gen XIST en una de las copias en las células trisómicas (Figure 7A).
11a.- Positive-negative selectable markers on the extra chromosome; the thymidine kinase-neomycin
(TKNEO) transgene is used, which helps to select with antibiotics and then isolate disomic cells from a
trisomic population. The fully trisomic cells (iPSCs-inducible pluripotent stem cells) are infected with
an adeno-associated viral vector (AAV) containing a TKNEO transgene that confers neomycin (NEO)
resistance and sensitivity to ganciclovir. Due to imperfect efficiency, only some cells in the
population receive the TKNEO transgene. The cell population is treated with neomycin, after which the
cells that do not contain the TKNEO transgene are removed. The population containing the pure transgene
is proliferated to allow nondisjunction events to occur naturally. The cohort of disomic and trisomic
cells is then treated with ganciclovir (GCV); all trisomic and TKNEO transgene-containing cells are
removed leaving only the pure disomic population that can be isolated and proliferated (Figure 7B).
11b.- Drug-induced trisomic rescue; where trisomic cells (trisomy 21 and 18) are cultured with
ZSCAN4, which increases the number of euploid (normal) cells by 24%.
11c.- Artificial human chromosomes (HAC-human artificial chromosomes); known as minichromosomes,
which are used as "vectors". These are freely integrated into the cell cycle over time, which would have
the possibility of correcting deletions.
11d.- Induction of ring chromosome formation. In trisomic cells (iPSCs) LoxP is inserted into
the short and long arm of the chromosome through CRISPR-Cas9. Then the cells are treated with a
recombinase that induces the formation of ring chromosomes, then these cells replicate and lose the ring
chromosome naturally, restoring the disomic state.
11e.- Inhibition of the DYRK1A gene, it has been demonstrated that this gene is involved in the
physiopathology of the intellectual disability of Down syndrome. One of the drugs that has demonstrated
its efficacy and safety in adult patients (phase 2) with Down syndrome is epigallocatechin-3-gallate
(green tea extract), improving cognition, visual recognition memory, inhibitory control and adaptive
behavior(86,87).
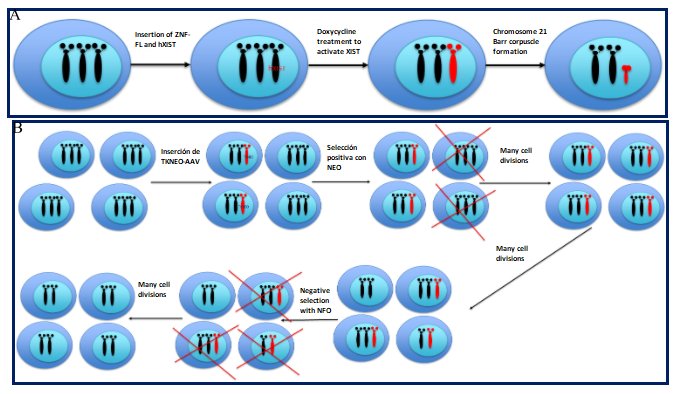 |
12. OTHER THERAPIES
Some monogenic diseases currently have therapies under clinical investigation, which could be verified
at www.clinicaltrials.com.
Some of the therapies shown are probably not directly focused on what is described in Figure 1;
however, they have been shown to have enormous utility in the management of these diseases.
Duchenne muscular dystrophy (DMD) is a pathology manifested by progressive loss of muscle
strength in the first decade. Three therapies are currently available, one of them we mentioned in short
nucleotide therapies, and the others are the use of deflazacort and ataluren. Deflazacort has been
widely used in DMD for more than 30 years; however, it was only approved by the FDA in 2017(88).
Ataluren is used in those patients who have a nonsense variant (10-15% of DMD patients). Its
mechanism of action is to perform a reading jump at the site of the nonsense variant, making the protein
larger than the "mutated" protein. In this way it causes the phenotype to change to Becker muscular
dystrophy(89).
In this same sense, the use of bisphosphonates in diseases such as osteogenesis imperfecta
(PS166200) and McCune-Albright syndrome (MIM #174800) are indicated to reduce pain and the risk of the
appearance of fractures(90,91).
Other therapies in osteogenesis imperfecta that have been observed to decrease the risk of
fractures is through the activation of osteoclasts (denosumab), bone anabolic agents (teriparatide,
romosozumab)(67).
X-linked hypophosphatemic rickets (MIM #307800) is a condition in which chronic hypophosphatemia
is observed which causes a failure in mineralization leading to rickets and osteomalacia. A monoclonal
FGF23 inhibitor (burosumab) has been shown to be a promising therapy in this condition(92).
Tuberous sclerosis (MIM #PS191100) has very heterogeneous clinical manifestations and the FDA
has approved the use of mTOR inhibitors such as everolimus for epilepsy, rhabdomyosarcomas,
astrocytomas, angiomyolipomas; and rapamycin for lymphangioleiomyomatosis(93).
Conclusions
The pharmacopoeia in genetic diseases is increasing notably over time. Many therapies try to be very
specific; however, drugs are being developed that will be used in more than one entity, which are even
etiologically unrelated.
As we are seeing in recent years, these new therapies are changing the natural history of this
group of entities. However, the bottleneck in these conditions is diagnosis, either due to the limited
number of specialists, lack of implementation, high costs, insurance coverage, among others.
The future of medicine in general is conditioned to better understand the underlying and
inherent mechanisms of each disease, based on an individual understanding of our "omics", thus taking it
to another level of medicine: Precision Medicine.
Finally, it is important to indicate that all these therapies and drugs are promising and
valuable therapeutic options for these different diseases described; however, it is of utmost importance
that the management of all these conditions is multi and interdisciplinary and carried out by qualified
professionals within laboratories and institutions properly certified for these purposes.
Authorship contributions: All authors have participated in the research design, data
collection and manuscript writing.
Funding Sources: None
Conflicts of interest: The authors have not declared any conflict of interest.
Received: April 7, 2020
Approved: December 1, 2020
Correspondence: Hugo Hernán Abarca Barriga.
Address: Servicio de Genética & EIM, Instituto Nacional de Salud del Niño, Av.
Brasil 600, CP Lima 05, Lima, Perú.
Telephone: +51 979301132
E-mail: habarca@insn.gob.pe
BIBLIOGRAPHIC REFERENCES
