CASO CLÍNICO
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2022 - Universidad Ricardo Palma10.25176/RFMH.v22i2.3816
NEFROMA MESOBLÁSTICO CONGÉNITO: REPORTE DE CASO
CONGENITAL MESOBLASTIC NEPHROMA: CASE REPORT
Carolina F. Paz Soldán Mesta1,a, José Luis Apaza León2,b, Mariela Tello Pezo3,c, Roxana M. Lipa Chancolla3,d
1 Unidad de Cirugía Pediátrica y Neonatal del Instituto Nacional de Salud San Borja Lima, Perú.
2 Unidad de Cirugía Pediátrica y Neonatal del Instituto Nacional de Salud San Borja. Lima, Perú
3 Instituto Nacional de Salud San Borja. Lima, Perú
a Cirujana Pediatra
b Jefe de la Unidad de Cirugía Pediátrica
c Oncóloga Pediatra
d Anatomopatóloga
RESUMEN
El nefroma mesoblástico congénito (NMC), es el tumor renal más frecuente en recién nacidos e infantes menores de 3 meses. Se presenta el caso clínico de una paciente menor de 3 meses, con diagnóstico prenatal, referido de manera oportuna para evaluación y manejo la Institución. El NMC es un tumor de baja incidencia. El diagnóstico precoz así como la excéresis completa del tumor son predictores de buen pronóstico, como en el caso de nuestra paciente.
Palabras Clave: Nefroma mesoblástico congénito; tumor renal; lactantes. (Fuente: DeCS BIREME).
ABSTRACT
Keywords: (Source : MeSH - NLM).
INTRODUCCIÓN
El nefroma mesoblástico congénito (NMC), también llamado hamartoma renal fetal, hamartoma leiomiomatoso
o mesenquimal, es un tumor de buen pronóstico, a excepción de la estirpe celular(1). Descrito por primera vez por Bolande en 1967, como un tumor distinto al
nefroblastoma por su histología, tratamiento y pronóstico(2). Representa el 3
a 5% de todos los tumores renales en pediatría(3), con mayor frecuencia en
recién nacidos e infantes menores de 3 meses(2). La recurrencia local y
metástasis es de 5% en el primer año(4).
El origen es probablemente por la proliferación nefrogénica mesenquimal(5). El NMC tiene una sobrevida de 85% a los 5 años. La metástasis es local con
mayor frecuencia, seguida de compromiso pulmonar, hepático, cerebral y cardiaca. La sobrevida después de
la recurrencia o con metástasis es de 57% a los 5 años(6).
CASO CLÍNICO
Se presenta el caso clínico de una lactante de 54 días de vida, nacida de parto a término, sin
antecedentes, procedente de Ancash. Referida a la institución con diagnóstico de masa renal derecha en
la ecografía prenatal durante el tercer trimestre. La madre es una mujer joven sin antecedentes de
importancia ni hábitos nocivos.
En la evaluación física de la paciente se palpa una masa en flanco derecho, no móvil ni
dolorosa. Los laboratorios no mostraron alteraciones, ni presentaba otra sintomatología. En la ecografía
se confirma una tumoración heterogénea de bordes regulares en 1/3 superior de riñón derecho. (Figura 1a)
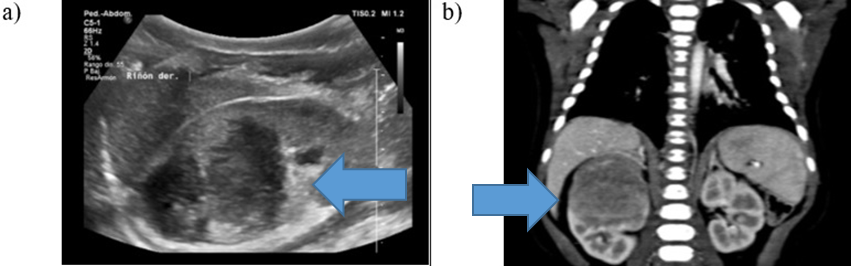
b) TAC abdominal con contraste: lesión expansiva de densidad discretamente heterogénea, de 41mm x 35mm x 36mm.
Se realizó una tomografía computarizada donde se evidencia una lesión expansiva de densidad
discretamente heterogénea, localizada en la mitad superior del riñón derecho, de 41mm x 35mm x 36mm (DL
x DAP x DT). La masa se encontraba confinada al parénquima renal, alcanzando la pelvis renal con leve
ectasia piélica y escasa vascularidad sin captación significativa del contraste ni calcificaciones
internas en el tumor. El riñón derecho presenta una variante anatómica con dos venas renales derechas.
(Figura 1b)
Se presentó en el Comité de Oncología de la Institución y se decidió realizar nefrectomía
radical derecha, extrayéndose la pieza sin complicaciones, siendo dada de alta a los 9 días de la
cirugía. En el servicio de anatomía patológica se recibe el riñón derecho, glándula suprarenal derecha y
uréter derecho. La tumoración mide 4.2cm x 3.9cm x 3cm, localizado en 2/3 superiores de riñón derecho,
de color blancogrisaceo, aspecto fibroso y mixoide, sin necrosis, ni hemorragia, representando el 50%
del volumen renal derecho.
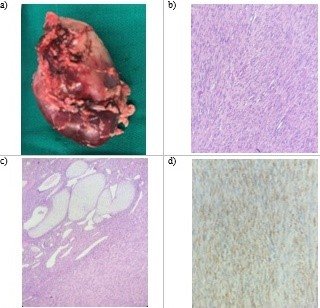
b) Neoplasia con células fusiformes delgadas dispuestas en fascículos que se entrecruzan.
c) Presencia de islas de tejido cartilaginoso, glomérulos y células fusiformes.
d) Inmunohistoquímica positivo a Actina músculo liso.
Figura 2: Estudio anatomopatológico de tumor renal.
La microscopía informó NMC, variante clásico, con infiltración de cápsula renal, grasa perirenal, pelvis
renal y fascia de gerota. La vena renal, seno renal y borde quirúrgico del uréter se encontraban libre
de neoplasia. La inmunohistoquímica informa WT-1 y Desmina negativa, actina y vimentina positiva y Ki-67
de 2%. (Figura 2)
La paciente no recibió quimioterapia neoadyuvante ni coadyuvante y es controlada en consultorio
externo, sin presentar complicaciones quirúrgicas ni recidiva, con una sobrevida hasta el momento mayor
de 3 años.
DISCUSIÓN
Aproximadamente el 5% de los tumores perinatales surgen del riñón. El NMC es el segundo tumor en
frecuencia después del tumor de Wilms durante el primer año y es el más común en los primeros 6 meses de
edad,(2) como lo fue el caso de la paciente con 2 meses de vida. A diferencia
de los publicado por Geramizadeh, con pacientes de edad entre los 18 meses a 11 años(7).
Existen 3 tipos histológicos: clásico (24%), celular (66%) y mixto (10%), siendo el clásico de
mejor pronóstico(4). Intraútero se manifiesta con polihidramnios e hidrops
fetal, asociado a parto prematuro e hipercalcemia(5). E cuadro clínico se
presenta como masa palpable en flanco (31.8%), hematuria (27.3%), dolor lumbar (22.7%) e hipertensión
por aumento de renina(8), puede presentar hipertensión pulmonar y falla
cardiaca(9).
Es más frecuente en el sexo femenino(8), difiriendo a los resultados
encontrados por Pachls, donde predominaba el sexo masculino(10). El
diagnóstico diferencial es con el tumor de Wilms, el tumor estromal metanéfrico y el sarcoma de células
claras del riñón, cursando con peor pronóstico(1). La ecografía obstétrica
permite el diagnóstico intraútero(2). Mientras que la tomografía axial
computarizada de abdomen permite realizar el diagnóstico, así como diferenciar entre los tipos
histológicos(5).
El tratamiento inicial es quirúrgico a excepción de aquellos pacientes con alta sospecha de
histología celular y mayores de 3 meses, donde la quimioterapia está indicada desde el
preoperatorio(4). Asimismo, cuando los tumores son de mayor tamaño, la
quimioterapia permite una reducción, con menor riesgo de complicaciones en el intraoperatorio(2). La quimioterapia coadyuvante está indicada en aquellos pacientes con
resección tumoral incompleta o ruptura del tumor durante su excéresis, así como en recurrencia local y
metástasis.
Existen 3 variantes histológicas del NMC: clásica, celular y mixta. El tipo clásico se
caracteriza por ser un tumor sólido y firme con presencia de cápsula y células fibroblásticas, con baja
actividad mitótica y abundante depósito de colágeno. El tipo celular con largas áreas hemorrágicas con
componente necrótico, alta actividad mitótica, invasión de grasa periférica y tejido conectivo(11). En la inmunohistoquimica, el NMC cursa con vimentina y actina de tejido
liso positivo, la desmina y CD 34 negativa(12).
La anatomía patológica de la paciente informó histología tipo clásica con bordes quirúrgicos
libres de tumor. Actualmente la paciente se encuentra libre de enfermedad, sin recurrencia luego de 3
años de la cirugía. A diferencia de lo publicado por Jehangir, con una recurrencia de hasta 71% y
metástasis de 42% en paciente con histología tipo celular, siendo este un factor de riesgo(13).
Los factores de mal pronósticos reportados son la edad mayor de 3 meses, bordes quirúrgicos
positivos, variante histológico celular y ruptura del tumor durante su excéresis(14). La paciente no presentó ninguno de estos factores.
CONCLUCIONES
El NMC es un tumor de baja incidencia, más frecuente en recién nacidos y lactantes menores de 3 meses.
Los factores de buen pronóstico permiten una mayor sobrevida. El tratamiento inicial es quirúrgico, a
excepción de pacientes que presentan factores de riesgo.
La evaluación integral, identificar los signos de alarma y la intervención precoz, son factores
que favorecerán la sobrevida en pacientes pediátricos con tumor sólido abdominal, es por ello la
importante de conocer esta patología.
Contribuciones de autoría: Carolina Paz Soldán Mesta y José Luis Apaza León participaron
en la concepciólo, recolección del artículo, recolección de datos y aprobación de la versión
final. Carolina Paz Soldán realizo el análisis de datos. Mariela Tello y Roxana Lipa realizaron
el aporte de paciente, redacción del artículo, aporte de material de estudio. Carolina Paz
Soldán realizo a revisión crítica del artículo y aprobación final.
Financiamiento: Autofinanciado.
Conflicto de intereses: Los autores declaran no tener conflictos de intereses.
Recibido: 01 de mayo 2021
Aprobado: 16 de febrero 2022
Correspondencia: Carolina Fabiola Paz Soldán Mesta
Dirección: Avenida Javier Prado Oeste 555, Dpto 103. San Isidro.
Teléfono: 961788484
Correo: carolina_pazsoldan_mesta@hotmail.com
REFERENCIAS
