CASO CLÍNICO
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2022 - Universidad Ricardo Palma10.25176/RFMH.v22i3.5007
XANTOGRANULOMA JUVENIL: ENFOQUE CLÍNICO-PATOLÓGICO. REPORTE DE DOS CASOS
JUVENILE XANTHOGRANULOMA: CLINICAL-PATHOLOGICAL APPROACH.CASE REPORTS
Eugenio Américo Palomino Portilla 1,2,3, Tula Ayquipa Arróspide 5 , Isaira Giovanna Torpoco Baquerizo 2,3, María Angélica Medrano Huallanca 2,4 , María del Pilar Quiñones Ávila 1,3
1Médico anatomopatólogo, Servicio de Patología Quirúrgica, Hospital Rebagliati
2Docente de Patología, Facultad de Medicina, Universidad Ricardo Palma
3Médico anatomopatólogo en Laboratorio Diagnosis S.A.C.
4Médico anatomopatólogo en Hospital Emergencia Ate-Vitarte
5Médico residente de Anatomía patológica, Facultad de Medicina Humana Universidad Ricardo Palma
RESUMEN
El Xantogranuloma juvenil es una lesión no neoplásica cutánea del tipo histiocitosis no-Langerhans, que afecta población principalmente pediátrica y usualmente de curso autolimitado. Excepcionalmente, la lesión es multifocal, ocular e incluso visceral, causando severas complicaciones. Se presenta el caso de dos pacientes de sexo femenino femeninas de 10 años, sin ninguna otra sintomatología ni antecedentes importantes, ambas con lesión única, una en el muslo, de 3 meses de evolución, la otra en cuero cabelludo, de 4 meses de evolución, ambas con crecimiento progresivo y extirpadas quirúrgicamente. Al estudio anatomopatológico se identificó múltiples histiocitos en dermis, con lipidización citoplasmática y formando células gigantes multinucleadas, algunas de tipo Touton, características de ésta lesión. Se revisa esta entidad inusual, con énfasis en los criterios histopatológicos y el curso clínico habitual.
Palabras Clave: Xantogranuloma; histiocitos; lipidización; Touton; reporte de casos. (fuente: DeCS BIREME).
ABSTRACT
Juvenile Xanthogranuloma is a non-neoplastic skin lesion of the non-Langerhans histiocytosis type, which mainly affects the pediatric population and usually has a self-limited course. Exceptionally, the lesion is multifocal, ocular and even visceral, causing severe complications. We present the case of two 10-year-old female patients, with no other symptoms or important history, both with a single lesion, one on the thigh, with 3 months of evolution, the other on the scalp, with 4 months of evolution, both with progressive growth and surgically removed. The anatomopathological study identified multiple histiocytes in the dermis, with cytoplasmic lipidization and forming giant multinucleated cells, some of the Touton type, characteristic of this lesion. This unusual entity is reviewed, with emphasis on the histopathological criteria and the usual clinical course.
Keywords: Xanthogranuloma; Histiocytes; Lipidization; Touton; case reports. (fuente: MeSH NLM).
INTRODUCCIÓN
El Xantogranuloma juvenil (XJ) es una lesión no neoplásica de histiocitos que acumulan colesterol en su citoplasma y pertenece al grupo de las Histiocitosis de células No-Langerhans (1). Suele afectar la piel, con curso autolimitado, no siendo necesaria la extirpación quirúrgica ni algún tratamiento.
Ocasionalmente compromete órganos internos, sobre todo cuando los nódulos cutáneos son múltiples y, más raro aún, sin ningún compromiso cutáneo (2). El compromiso ocular es uno de los más comunes, causando glaucoma, sangrado y hasta ceguera (3).
La afección predomina en la infancia, entre los 5 y 12 meses (4) aunque se reporta una forma congénita (5). La relación varón/mujer es en promedio de 1.4:1, sin predilección por grupos étnicos (6).
La patogenia favorece un origen reactivo mas que neoplásico, en donde un daño inespecífico generaría una respuesta macrofágica desordenada (7). Estos macrófagos derivarían de los dendrocitos dermales y causarían el infiltrado dermal mixto de histiocitos, células gigantes mononucleares y hasta células fusiformes (8).
Clínicamente, suele ser una lesión solitaria, rojiza/amarillenta, en forma de placa, pápula o nódulo, que mide entre 5-20 mm y se ubica generalmente en cabeza, cuello, tronco, aunque puede afectar cualquier zona anatómica. Las formas extracutáneas se reportan en pulmón, hígado, bazo, ganglios linfáticos, tubo gastrointestinal, corazón, riñones, médula ósea, sistema nervioso central y ojos. En el caso de la forma cutánea típica, la regresión espontánea es la regla y toma entre 1 a 5 años, por lo cual no requiere tratamiento (9).
El diagnóstico suele ser fácil en las formas clínicas cutáneas típicas. Sin embargo, en las heterogéneas formas extracutáneas/viscerales, puede ocurrir error diagnóstico. La confirmación ante la sospecha clínica puede realizarse con el estudio patológico de una biopsia de piel y uso de un panel de inmunohistoquímica (10).
Al estudio patológico, microscópicamente, se observa en la dermis e hipodermis superior un infiltrado denso, en sábana, bien delimitado y no encapsulado. La epidermis con sus anexos cutáneos suele ser comprimida. El infiltrado suele estar compuesto por 5 tipos de células (vacuoladas, xantomatosas, fusiformes, festoneadas y oncocíticas) en proporciones diversas. Los núcleos de los histiocitos son redondos u ovales, el citoplasma finamente vacuolado, espumoso. Hay diferentes tipos de células gigantes: inespecíficas, a cuerpo extraño, tipo Touton y otras en vidrio esmerilado. El aspecto general varía con la edad de la lesión: lesiones tempranas contienen macrófagos monomorfos sin lípidos, en cambio lesiones maduras contienen abundantes macrófagos vacuolados, xantomatosos y particularmente, células de Touton (10), las cuales son células gigantes multinucleadas que al corte presentan un anillo de 10 o más núcleos, dividiendo al citoplasma en una porción central homogénea, eosinófila (rica en mitocondrias y lisosomas) y en una región periférica, espumosa (rica en material lipídico). Las células de Touton son comunes en el XJ, pero no son patognomónicas. También se observan neutrófilos, eosinófilos, linfocitos y, rara vez, mastocitos, en ocasiones con fibrosis (11). La cantidad de fibrosis es variable, a veces formando un vago patrón estoriforme (12).
Se han descrito tres aspectos histológicos. La forma temprana consta de histiocitos con poca o nula lipidización, linfocitos y eosinófilos. La forma clásica presenta histiocitos vacuolados, con lipidización y células gigantes de Touton. La forma transicional presenta fibrosis y se observa células fusiformes que recuerdan al histiocitoma fibroso benigno (4).
A diferencia de los macrófagos, las células de Langerhans suelen ser ovales o elípticas, con núcleo plegado, clivado y nucléolo poco notorio. Tampoco acumulan lípidos en su citoplasma (13).
Al estudio inmunohistoquímico las lesiones son positivas a marcadores de macrófagos (CD68, CD163, KiM1P, anti-FXIIIa, vimentina, anti-CD4) y son negativas para S-100, CD1a y CD207 (antilangerina), marcador específico para las células de Langerhans(14).
El diagnóstico diferencial histopatológico debe descartar Histiocitosis de células de Langerhans (ausencia de células de Touton, histiocitos positivos para CD1a, S100, gránulos de Birbeck a la microscopía electrónica, negatividad para CD68 y Factor XIII). También frente al Histiocitoma fibroso benigno (patrón estoriforme, bandas gruesas de colágeno, epidermis hiperplásica, CD34 y Factor XIIIa positivos). Igualmente, con Xantomas (ausencia de células de Touton, proliferación de macrófagos espumosos). Considerar también al Reticulohistiocitoma caracterizado por células grandes con citoplasma en vidrio esmerilado con núcleos al azar, junto con histiocitos y fibroblastos (15). El XJ no suele presentar emperipolesis (células intactas dentro del citoplasma de otra célula), no obstante se ha reportado un caso con éste detalle (16).
El Xantogranuloma juvenil y la Leucemia mielomonocítica juvenil pueden ser difíciles de diferenciar, clínica e histopatológicamente(17), lo cual amerita precisar el diagnóstico.
El XJ es una lesión cutánea típica, pero poco común. Además casi siempre es cutánea, pero cuando es visceral conlleva severa morbilidad, por lo cual se justifica un adecuado conocimiento clínico-patológico.
DESCRIPCIÓN DEL CASO CLÍNICO
Ambos casos comparten las características histopatológicas típicas del XJ, el perfil inmunohistoquímico y la carencia de recidiva, pero difieren del cuadro común en la edad poco usual de presentación, en el género femenino y en que fueron tratadas con extirpación quirúrgica, detalle poco común con un diagnóstico clínico apropiado.
CASO 1. Paciente de sexo femenino, de 10 años de edad, sin antecedentes de importancia, presentó lesión cutánea nodular en muslo derecho, color amarillento, de 3 meses de evolución y crecimiento lentamente progresivo, no asociado a dolor ni a síntomas constitucionales. Dado que la lesión incrementó su tamaño, el médico decidió realizar la extirpación de la lesión.
Se realizó el examen patológico, en donde macroscópicamente, se recibe un losange de piel, fijado en formol, de 8x7x6 milímetros, color pardusco, en cuya superficie presenta lesión nodular, de 6x4x4 milímetros, amarillenta, consistencia elástica. Al corte, la superficie es sólida, amarillenta, ovalada, de contornos definidos. Al estudio microscópico, la epidermis está adelgazada, con hiperqueratosis difusa (Figura 1, A), daño focal por inflamación crónica asociada a célula gigante multinucleada tipo cuerpo extraño. No se reconoce anexos cutáneos en la lesión. La dermis muestra nódulo ovalado, de bordes bien definidos, no encapsulado que eleva la epidermis y la atrofia. La población celular corresponde principalmente a histiocitos, con y sin vacuolas. Dispersas entre ellos, se encuentran células gigantes multinucleadas tipo cuerpo extraño, tipo Langhans y tipo Touton, éstas últimas muy características, con un anillo paracentral de núcleos delimitando un centro eosinófilo homogéneo y un área periférica intensamente vacuolada (Figura 1, C). Algunas células gigantes multinucleadas se ubican justo bajo la epidermis y focalmente se identifica emperipolesis. Los histiocitos y células gigantes predominan en la periferie. Además se reconoce linfocitos, escasas células plasmáticas y eosinófilos. El centro de la lesión presenta colágeno y fibroblastos, con vago patrón estoriforme (Figura 1, A), en medio del cual la población histiocitaria y de células gigantes multinucleadas es menor. La vascularización es una red de vasos de pequeño calibre. El panel de inmunohistoquímica realizado confirma reactividad de la población histiocitaria y de células gigantes para CD68 (Figura 1, F) y Factor XIIIa, así como no reactividad para proteína S100, CD1a y CD207. La evolución posquirúrgica cursó con cicatrización adecuada, sin ninguna complicación local ni sistémica.
CASO 2. Paciente de sexo femenino, de 10 años de edad, sin antecedentes de importancia, presentó lesión cutánea nodular en cuero cabelludo, región fronto-parietal izquierda, color amarillento, de 4 meses de evolución y crecimiento lentamente progresivo, con prurito local. No se asocia a dolor ni síntomas constitucionales. Dado que la lesión incrementó su tamaño, el médico hace el diagnóstico clínico de nevus parietal y realiza la exéresis de la lesión.
Se realizó el examen patológico, en donde macroscópicamente se recibe un losange de piel, fijado en formol, de 7x6x5 milímetros, color pardusco, en cuya superficie se observa lesión nodular, de 6x5x4 milímetros, amarillenta, consistencia elástica. Al corte, la superficie es homogénea, amarillenta, ovalada, de contornos definidos. Al estudio microscópico, la epidermis es atrófica, sin hiperqueratosis ni daño focal por inflamación. Se observa folículos pilosos inmersos en la lesión (Figura 1, B). La dermis presenta un nódulo ovalado, de bordes bien definidos, no encapsulado, que eleva la epidermis y la atrofia. La población celular corresponde principalmente a histiocitos, con vacuolización notoria. Dispersos entre ellos, se encuentra células gigantes multinucleadas tipo cuerpo extraño, tipo Langhans y tipo Touton (Figura 1, D). Algunas células gigantes multinucleadas se ubican justo bajo la epidermis. No se identifica emperipolesis. La población histiocitaria y de células gigantes predomina en la periferie. Semejante al caso anterior, se observa linfocitos, escasas células plasmáticas y eosinófilos. Toda la lesión es celular (Figura 1, B), sin fibrosis central. La vascularización es una red de vasos de pequeño calibre. Semejante al caso 1, el panel de inmunohistoquímica realizado confirma positividad de la población histiocitaria y de células gigantes para el CD68 y Factor XIIIa, así como negatividad para proteína S100, CD1a y CD207 (Figura 1, F). La evolución posquirúrgica cursó con cicatrización adecuada, sin ninguna complicación local ni sistémica.
En ambos casos, las láminas histopatológicas y bloques de tejido fueron obtenidos del archivo institucional, meses después de los procedimientos quirúrgicos. Igualmente, los datos clínicos fueron obtenidos de la información clínica archivada. La identificación de las pacientes siempre se ha mantenido en la reserva debida.
FIGURA 1.
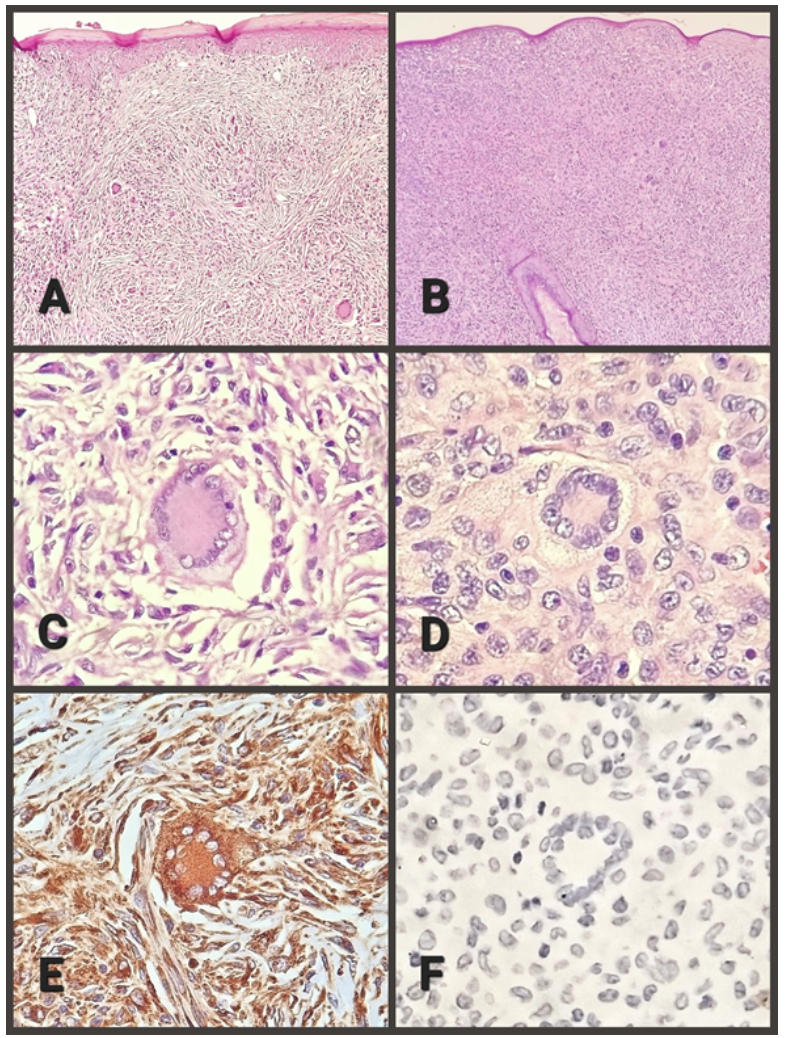
B. Caso 2, ausencia de fibrosis, celularidad notoria y persistencia de folículo piloso (100X).
C. Caso 1, célula de Touton y macrófagos (400X).
D. Caso 2, célula de Touton y macrófagos (400X).
E. Caso 1, célula de Touton y macrófagos con inmunorreacción positiva para CD-68 (400X).
F. Caso 2, célula de Touton y macrófagos con inmunorreacción negativa para CD-207 (400X).
DISCUSIÓN
Los dos casos reportados presentan limitaciones como la aparición en edad, género y ubicación inusuales, que conllevaron a un diagnóstico clínico erróneo y a la extirpación quirúrgica. Sin embargo, el estudio del espécimen quirúrgico generó las fortalezas, como el cuadro histopatológico típico y la confirmación del diagnóstico por medio de anticuerpos de inmunohistoquímica.
El Xantogranuloma juvenil posee características clínico-patológicas particulares, la mayoría de las cuales están presentes en los dos casos presentados. La edad de aparición más común es el primer año de vida, pudiendo presentarse incluso en adultos. Coincidentemente, los dos casos reportados aparecieron a los 10 años. Respecto al género, se describe leve predominancia masculina y los dos casos presentados correspondieron a niñas. En cuanto al origen de la lesión, en ninguno de los dos casos se reportó antecedentes de trauma o lesión local que sugieran una naturaleza reactiva.
Clínicamente, ambas son lesiones nodulares, menores de 1cm de diámetro, color amarillento, consistencia elástica, de lento crecimiento. El primer caso no reportó ningún malestar asociado, mientras que en el segundo caso se reportó prurito local.
El estudio microscópico identificó una lesión no encapsulada, de bordes bien definidos, que eleva y atrofia la epidermis en ambos casos, con hiperqueratosis y daño focal inflamatorio solo en el primer caso. Los anexos cutáneos no se identifican en el caso 1 y si se reconocen en el caso 2. La población celular diagnóstica es muy característica en ambos casos: histiocitos, con lipidización o nó, células gigantes multinucleadas tipo cuerpo extraño y tipo Langhans, destacando las muy características, pero no patognomónicas, células de Touton. En el caso 1 se identificó emperipolesis, mientras en el segundo caso, no. Las células inflamatorias acompañantes son linfocitos, plasmocitos y eosinófilos dispersos en ambos casos. El caso 1 presenta fibrosis notoria con aspecto estoriforme (patrón transicional), mientras que el caso 2 carece de fibrosis.
La reactividad de la población histiocitaria con los anticuerpos de inmunohistoquímica corresponde a lo descrito en la literatura: inmunorreactividad positiva frente a CD68 y Factor XIIIa, mientras que para proteína S100, CD1a y CD207 hay carencia de reactividad. El Cuadro 1 resume las características clínico-patológicas de ambos casos.
Un detalle interesante es que ambos casos fueron atendidos inicialmente por médicos cirujanos, no pediatras, quienes procedieron a la extirpación quirúrgica con un diagnóstico de tumoración de piel en el primer caso y de nevus parietal en el segundo caso. En ambas pacientes, no hubo recurrencia local de la lesión ni aparición en otro lugar.
CUADRO 1
CARACTERÍSTICAS CLÍNICO-PATOLÓGICAS
| CASO 1 | CASO 2 | ||
|---|---|---|---|
| Ubicación | Muslo derecho | Frontoparietal izquierda | |
| Color superficial | Amarillento | Amarillento | |
| Tiempo de evolución | Tres meses | Cuatro meses | |
| Velocidad de crecimiento | Lenta | Lenta | |
| Síntomas | Ninguno | Prurito local | |
| Hallazgos macroscópicos | Tamaño | 6x4x4 mm | 6x5x4 mm |
| Color al corte | Amarillento | Amarillento | |
| Forma | Ovalada | Ovalada | |
| Bordes | Bien definidos | Bien definidos | |
| Hallazgos microscópicos | Atrofia epidérmica | Si | Si |
| Inflamación epidérmica | Si | No | |
| Hiperqueratosis | Si | No | |
| Anexos cutáneos | Ausentes | Presentes | |
| Histiocitos con/sin lipidización | Presentes | Presentes | |
| Células gigantes multinucleadas | Presentes | Presentes | |
| Células de Touton | Presentes | Presentes | |
| Emperipolesis | Si | No | |
| Linfocitos/plasmocitos/eosinófilos | Presentes | Presentes | |
| Fibrosis | Presente | Ausente | |
| Vascularización | Pequeño calibre | Pequeño calibre | |
| Inmunorreacción positiva | CD68, Factor XIIIa | CD68, Factor XIIIa | |
| Inmunorreacción negativa | S100, CD1a, CD207 | S100, CD1a, CD207 | |
| Recurrencia | No | No | |
La ubicación anatómica de la lesión en el caso 1 es infrecuente, ambos casos aparecieron a una edad inusual, con hallazgos macro y microscópicos característicos, al igual que el clásico perfil de inmunohistoquímica. Ambas fueron extirpadas quirúrgicamente, lo cual permitió el examen anatomopatológico definitivo.
CONCLUSIÓN
Los casos reportados permiten identificar la histopatología clásica de la lesión, tras un diagnóstico clínico que decidió la extirpación. En otro contexto, una evaluación pediátrica suele llegar al diagnóstico clínico y, conociendo su curso autolimitado, se evita la cirugía
Otro detalle que justifica conocer mejor al XJ, es la posibilidad de coexistencia entre lesiones cutáneas y viscerales, sobre todo a nivel ocular, con pronóstico y tratamiento muy diferentes. Se justifica entonces, la búsqueda de lesiones extracutáneas del XJ.
Finalmente, se han descrito semejanzas clínico-patológicas entre el Xantograuloma juvenil y la Leucemia mielomonocítica juvenil, entidades completamente diferentes, por lo cual es vital alcanzar un diagnóstico confirmado de la entidad descrita.
Contribuciones de Autoría: Eugenio Américo Palomino Portilla: redacción del artículo, aprobación de la versión final
Isaira Giovanna Torpoco Baquerizo: elaboración de las conclusiones del artículo
María Angélica Medrano Huallanca y Tula Dariela Ayquipa Arróspide: elaboración de la discusión del artículo
María del Pilar Quiñones Ávila: concepción y diseño del artículo, revisión crítica del artículo.
Financiamiento: Autofinanciado.
Conflictos de intereses: Los autores declaran no tener conflicto de interés.
Recibido: 01 de abril 2022
Aprobado: 02 de Junio 2022
Correspondencia: Eugenio Américo Palomino Portilla.
Dirección: Calle Doña Victoria, C-1, Surco.
Teléfono: 998 483195
Email: eugenio.palomino@urp.edu.pe
Artículo publicado por la Revista de la Facultad de Medicina Humana de la Universidad Ricardo Palma. Es un articulo de acceso abierto, distribuido bajo los términos de la Licencia Creatvie Commons: Creative Commons Attribution 4.0 International, CC BY 4.0(https://creativecommons.org/licenses/by/1.0/), que permite el uso no comercial, distribucion y reproducción en cualquier medio, siempre que la obra original sea debidamente citada. Para uso comercial, por favor póngase en contacto con revista.medicina@urp.edu.pe.
REFERENCIAS
