CASO CLÍNICO
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2022 - Universidad Ricardo Palma10.25176/RFMH.v22i3.5007
JUVENILE XANTHOGRANULOMA: CLINICAL-PATHOLOGICAL APPROACH. CASE REPORTS
XANTOGRANULOMA JUVENIL: ENFOQUE CLÍNICO-PATOLÓGICO. REPORTE DE DOS CASOS
Eugenio Américo Palomino Portilla 1,2,3, Tula Ayquipa Arróspide 5 , Isaira Giovanna Torpoco Baquerizo 2,3, María Angélica Medrano Huallanca 2,4 , María del Pilar Quiñones Ávila 1,3
1Médico anatomopatólogo, Servicio de Patología Quirúrgica, Hospital Rebagliati
2Docente de Patología, Facultad de Medicina, Universidad Ricardo Palma
3Médico anatomopatólogo en Laboratorio Diagnosis S.A.C.
4Médico anatomopatólogo en Hospital Emergencia Ate-Vitarte
5Médico residente de Anatomía patológica, Facultad de Medicina Humana Universidad Ricardo Palma
ABSTRACT
Juvenile Xanthogranuloma is a non-neoplastic skin lesion of the non-Langerhans histiocytosis type, which mainly affects the pediatric population and usually has a self-limited course. Exceptionally, the lesion is multifocal, ocular, and even visceral, causing severe complications. We present the case of two 10-year-old female patients, with no other symptoms or important history, both with a single lesion, one on the thigh, with 3 months of evolution, the other on the scalp, with 4 months of evolution, both with progressive growth and surgically removed. The anatomopathological study identified multiple histiocytes in the dermis, with cytoplasmic lipidization and forming giant multinucleated cells, some of the Touton type, characteristic of this lesion. This unusual entity is reviewed, with emphasis on the histopathological criteria and the usual clinical course.
Keywords: Xanthogranuloma; histiocytes; lipidization; Touton; case reports.(fuente: MeSH NLM).
RESUMEN
El Xantogranuloma juvenil es una lesión no neoplásica cutánea del tipo histiocitosis no-Langerhans, que afecta población principalmente pediátrica y usualmente de curso autolimitado. Excepcionalmente, la lesión es multifocal, ocular e incluso visceral, causando severas complicaciones. Se presenta el caso de dos pacientes de sexo femenino femeninas de 10 años, sin ninguna otra sintomatología ni antecedentes importantes, ambas con lesión única, una en el muslo, de 3 meses de evolución, la otra en cuero cabelludo, de 4 meses de evolución, ambas con crecimiento progresivo y extirpadas quirúrgicamente. Al estudio anatomopatológico se identificó múltiples histiocitos en dermis, con lipidización citoplasmática y formando células gigantes multinucleadas, algunas de tipo Touton, características de ésta lesión. Se revisa esta entidad inusual, con énfasis en los criterios histopatológicos y el curso clínico habitual.
Palabras Clave: Xantogranuloma; histiocitos; lipidización; Touton; reporte de casos. (fuente: DeCS BIREME).
INTRODUCTION
Juvenile Xanthogranuloma (JXG) is a non-neoplastic lesion of histiocytes that accumulate cholesterol in their cytoplasm and belongs to the group of non-Langerhans cell histiocytosis (1). It usually affects the skin, with a self-limited course, not requiring surgical removal or any treatment.
Occasionally it involves internal organs, especially when the skin nodules are multiple and, even more rarely, without any skin involvement (2). Ocular involvement constitutes one of the most common clinical presentations, causing glaucoma, bleeding, and even blindness (3).
It predominates in childhood, between 5 and 12 months (4), although a congenital form has been described (5). The male/female ratio is on average 1.4:1, with no predilection for ethnic groups (6).
The pathogenesis favors a reactive rather than neoplastic origin, where nonspecific damage would generate a disordered macrophage response (7). These macrophages would derive from dermal dendrocytes and cause a mixed dermal infiltrate of histiocytes, mononuclear giant cells, and even spindle cells (8).
Clinically, it is usually a solitary, reddish/yellowish lesion, in the form of a plaque, papule, or nodule, measuring between 5-20 mm and generally located on the head, neck, trunk, although it can affect any anatomical area. Extracutaneous forms are reported in the lung, liver, spleen, lymph nodes, gastrointestinal tract, heart, kidneys, bone marrow, central nervous system, and eyes. In the case of the typical cutaneous form, spontaneous regression is the rule and lasts between 1 to 5 years, so it does not require treatment (9).
Diagnosis is usually easy in the typical cutaneous clinical forms. However, in heterogeneous extracutaneous/visceral forms, misdiagnosis can occur. Confirmation of clinical suspicion can be done with the pathological study of a skin biopsy and the use of an immunohistochemical panel (10).
In the anatomopathological study, under the microscope, a dense, well-defined, and non-encapsulated infiltrate is observed in the upper dermis and hypodermis. The epidermis with its skin appendages is usually compressed. In variable proportions, the infiltrate is usually composed of 5 cell types (vacuolated, xanthomatous, spindle-shaped, scalloped, and oncocytic). Histiocyte nuclei are round or oval, the cytoplasm is finely vacuolated and foamy. There are different types of giant cells: nonspecific, foreign body, Touton type, and ground glass. The general appearance varies with the age of the lesion: early lesions contain lipid-free monomorphic macrophages, whereas mature lesions contain abundant vacuolated xanthomatous macrophages and, in particular, Touton cells (10), which are multinucleated giant cells that when cut, they present a ring of 10 or more nuclei, which divides the cytoplasm into a homogeneous central portion, eosinophilic (rich in mitochondria and lysosomes) and a peripheral foamy region (rich in lipid material). Touton cells are common in JXG but are not pathognomonic. Neutrophils, eosinophils, lymphocytes, and rarely mast cells, sometimes with fibrosis, are also seen (11). The amount of fibrosis is variable, sometimes forming a vague storiform pattern (12).
Three histological aspects have been described. The early form consists of histiocytes with little or no lipidization, lymphocytes, and eosinophils. The classic form presents vacuolated histiocytes, with lipidization and giant Touton cells. The transitional form presents with fibrosis and fusiform cells reminiscent of benign fibrous histiocytoma (4).
Unlike macrophages, Langerhans cells are usually oval or elliptical, with folded and cleft nuclei and inconspicuous nucleoli. They also do not accumulate lipids in their cytoplasm (13).
In the immunohistochemical study, the lesions are positive for macrophage markers (CD68, CD163, KiM1P, anti-FXIIIa, vimentin, anti-CD4) and are negative for S-100, CD1a, and CD207 (antilangerin), a specific marker for Langerhans cells (14).
Histopathological differential diagnosis should rule out Langerhans cell histiocytosis (absence of Touton cells, histiocytes positive for CD1a, S100, Birbeck granules in electron microscopy, CD68 and Factor XIII negative). Also, Benign fibrous histiocytoma (storiform pattern, thick collagen bands, hyperplastic epidermis, CD34, and Factor XIIIa positive). Likewise, Xanthomas (absence of Touton cells, proliferation of foamy macrophages). Consider also reticulohistiocytoma characterized by large cells with ground glass cytoplasm and random nuclei, along with histiocytes and fibroblasts (15). JXG does not usually present emperipolesis (intact cells within the cytoplasm of another cell), however, a case with this detail has been reported (16).
Juvenile Xanthogranuloma and Juvenile Myelomonocytic Leukemia can be difficult to differentiate clinically and histopathologically (17), which requires an accurate diagnosis.
JXG is a typical but uncommon skin lesion. In addition, it is almost always cutaneous, but when it is visceral it carries severe morbidity, which is why adequate clinical-pathological knowledge is justified.
PRESENTATION OF CASES
Both cases share the typical histopathologic features of JXG, the immunohistochemical profile, and the absence of recurrence, but differ from the common picture in the unusual age of presentation, female gender, and in that they were treated with surgical excision, an unusual outcome with an appropriate clinical diagnosis.
CASE 1. A 10-year-old female patient, with no history of interest, presented a nodular skin lesion on the right thigh, yellowish in color, with a 3-month history and slowly progressive growth, not associated with pain or constitutional symptoms. Since the lesion increased in size, the doctor decided to remove the lesion.
The anatomopathological examination was performed, where macroscopically a lozenge of skin fixed in formalin was received, measuring 8x7x6 millimeters, brownish in color, on whose surface there is a nodular lesion, measuring 6x4x4 millimeters, yellowish in color, with an elastic consistency. When cut, the surface is solid, yellowish, and oval, with defined contours. In the microscopic study, the epidermis is thinned, with diffuse hyperkeratosis (Figure 1, A), and focal damage due to chronic inflammation associated with foreign body-type multinucleated giant cells. Skin appendages are not recognized in the lesion. The dermis shows a solid, well-defined, non-encapsulated nodule that elevates and atrophies the epidermis. The cell population corresponds mainly to histiocytes, with and without vacuoles. Scattered among them, there are giant multinucleated foreign-body, Langhans-type, and Touton-type cells, the latter very characteristic, with a paracentral ring of nuclei delimiting a homogeneous eosinophilic center and an intensely vacuolated peripheral area (Figure 1, C). Some multinucleated giant cells are located just below the epidermis and emperipolesis is focally identified. Histiocytes and giant cells predominate in the periphery. In addition, lymphocytes, few plasma cells, and eosinophils are recognized. The center of the lesion presents collagen and fibroblasts, with a vague storiform pattern (Figure 1, A), in the middle of which the population of histiocytic cells and multinucleated giant cells is smaller. Vascularization is a network of small caliber vessels. The immunohistochemical panel performed confirms the reactivity of the histiocytic and giant cell population for CD68 (Figure 1, F) and Factor XIIIa, as well as the non-reactivity for: protein S100, CD1a, and CD207. The post-surgical evolution was with adequate healing, without any local or systemic complications.
CASE 2. A 10-year-old female patient, with no relevant history, presented with a nodular skin lesion on the scalp, in the left frontoparietal region, yellowish in color, with a 4-month history and slowly progressive growth, with local itching. It is not associated with pain or constitutional symptoms. Since the lesion increased in size, the doctor makes a clinical diagnosis of parietal nevus and performs the exeresis of the lesion.
The pathological examination was performed, where macroscopically a lozenge of skin fixed in formalin, was received, measuring 7x6x5 millimeters, brownish in color, on whose surface a nodular lesion, 6x5x4 millimeters, yellowish, elastic consistency was observed. When cut, the surface is homogeneous, yellowish, oval, with defined contours. On microscopic study, the epidermis is atrophic, without hyperkeratosis or focal damage due to inflammation. Hair follicles immersed in the lesion are observed (Figure 1, B). The dermis presents a solid nodule, with well-defined borders, not encapsulated, which elevates and atrophies the epidermis. The cell population corresponds mainly to histiocytes, with marked vacuolization. Scattered among them, there are foreign-body-type, Langhans-type, and Touton-type multinucleated giant cells (Figure 1, D). Some multinucleated giant cells are located just below the epidermis. Emperipolesis not identified. Histiocytic and giant cell population predominates in the periphery. Similar to the previous case, lymphocytes, few plasma cells, and eosinophils are observed. The entire lesion is cellular (Figure 1, B), with no central fibrosis. Vascularization is a network of small caliber vessels. Similar to case 1, the immunohistochemical panel performed confirms the positivity of the histiocytic and giant cell population for CD68 and Factor XIIIa, as well as the negativity for protein S100, CD1a, and CD207 (Figure 1, F). Postoperative evolution was with adequate healing, without local or systemic complications.
In both cases, histopathological slides and tissue blocks were obtained from the institutional archive, months after the surgical procedures. Similarly, clinical data was obtained from archived clinical information. Patient identification has always been kept confidential.
Figure 1.
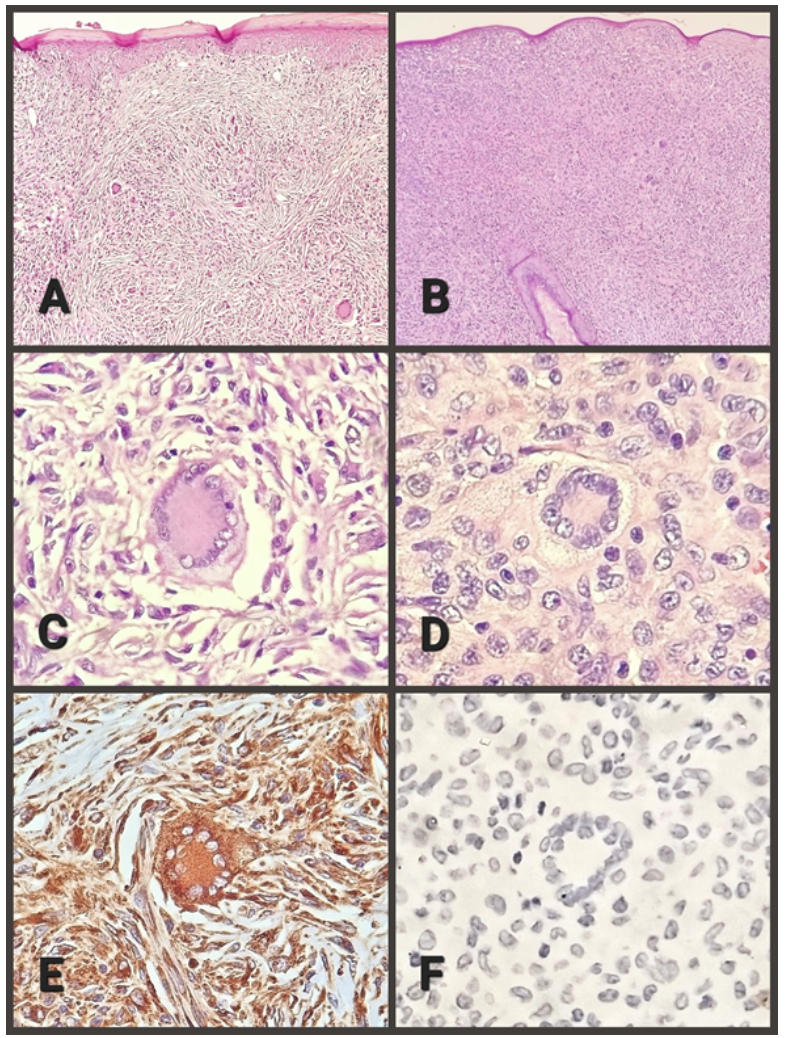
B. Case 2, absence of fibrosis, notable cellularity, and persistence of the hair follicle (100X).
C. Case 1, Touton cell and macrophages (400X)
D. Case 2, Touton cell and macrophages (400X).
E. Case 1, Touton cell and macrophages with positive immunoreaction for CD-68 (400X).
F. Case 2, Touton cell and macrophages with a negative immunoreaction for CD-207 (400X).
DISCUSSION
The two reported cases present limitations such as the appearance in unusual age, sex, and location, which led to a clinical misdiagnosis and surgical removal. However, the study of the surgical piece generated strengths, such as the typical histopathological picture and the confirmation of the diagnosis through immunohistochemical antibodies.
Juvenile Xanthogranuloma presents particular clinical-pathological characteristics, most of which are present in the two cases presented. The most frequent age of onset is the first year of life, but it can even occur in adults. Coincidentally, the two reported cases appeared at 10 years. Regarding gender, a slight male predominance is described and the two cases presented corresponded to girls. Regarding the origin of the injury, there was no history of trauma in any of the cases or local injury that would suggest a reactive nature.
Clinically, both are nodular lesions, less than 1 cm in diameter, yellowish in color, elastic in consistency, and slow growing. The first case reported no associated discomfort, while the second case reported local itching.
The microscopic study identified a non-encapsulated lesion with well-defined borders, that raised and atrophied the epidermis in both cases, with hyperkeratosis and focal inflammatory damage only in the first case. Skin appendages are not identified in case 1 and are recognized in case 2. The diagnostic cell population is very characteristic in both cases: histiocytes, with or without lipidization, foreign body type, and Langhans type multinucleated giant cells, highlighting the very characteristic, but not pathognomonic, Touton cells. In case 1, emperipolesis was identified, while in the second case, it was not.
The accompanying inflammatory cells are lymphocytes, plasma cells, and scattered eosinophils in both cases. Case 1 presents marked fibrosis with a storiform appearance (transitional pattern), while case 2 lacks fibrosis.
The reactivity of the histiocytic population with immunohistochemical antibodies corresponds to that described in the literature: positive immunoreactivity against CD68 and Factor XIIIa, while there is a lack of reactivity for protein S100, CD1a, and CD207.
Table 1 summarizes the clinicopathological characteristics of both cases.
An interesting detail is that both cases were initially treated by surgeons, not pediatricians, who proceeded to surgical removal with a diagnosis of skin tumor in the first case and parietal nevus in the second case. In both patients, there was no local recurrence of the lesion or appearance elsewhere.
Table 1
CLINICAL-PATHOLOGICAL CHARACTERISTICS
| CASE 1 | CASE 2 | ||
|---|---|---|---|
| Location | Right thigh | left frontoparietal | |
| Surface color | Yellowish | Yellowish | |
| Time of evolution | Three months | Four months | |
| Growth | Slow | Slow | |
| Symptoms | None | Local pruritus | |
| Macroscopic findings | Size | 6x4x4 mm | 6x5x4 mm |
| Color after cutting | Yellowish | Yellowish | |
| Form | oval | oval | |
| Borders | Well defined | Well defined | |
| Microscopic findings | Epidermal atrophy | Yes | Yes |
| Epidermal inflammation | Yes | No | |
| Hyperkeratosis | Yes | No | |
| Skin appendages | Missing | Present | |
| Histiocytes with/without lipidization | Present | Present | |
| Multinucleated giant cells | Present | Present | |
| Touton cells | Present | Present | |
| Emperipolesis | Yes | No | |
| Lymphocytes/plasma cells/eosinophils | Present | Present | |
| Fibrosis | Present | Missing | |
| Vascularization | Small caliber | Small caliber | |
| Positive immunoreaction | CD68, Factor XIIIa | CD68, Factor XIIIa | |
| Negative immunoreaction | S100, CD1a, CD207 | S100, CD1a, CD207 | |
| Recurrence | No | No | |
The anatomical location of the lesion in case 1 is unusual, both cases appeared at an unusual age, with characteristic macro and microscopic findings, as well as the classic immunohistochemical profile. Both were surgically removed, which allowed definitive pathological examination.
CONCLUSION
The reported cases allow us to identify the classic histopathology of the lesion, after a clinical diagnosis that decided the removal. In another context, a pediatric evaluation usually leads to a clinical diagnosis and, knowing its self-limited course, surgery is avoided.
Another detail that justifies a better understanding of JXG is the possibility of coexistence between cutaneous and visceral lesions, especially at the ocular level, with a very different prognosis and treatment. Therefore, the search for extracutaneous JXG lesions is justified.
Finally, clinical-pathological similarities have been described between juvenile xanthogranuloma and juvenile myelomonocytic leukemia, completely different entities, for which it is vital to achieve a confirmed diagnosis of the entity described.
Authorship contributions: Eugenio Américo Palomino Portilla: Writing of the article, approval of the final version.
Isaira Giovanna Torpoco Baquerizo: Elaboration of the conclusions of the article.
María Angélica Medrano Huallanca y Tula Dariela Ayquipa Arróspide: Elaboration of the discussion of the article.
María del Pilar Quiñones Ávila: Conception and design of the article, critical review of the article.
Funding sources: Self-financed.
Conflicts of interest: The authors declare no conflict of interest.
Received: May 16, 2022
Approved: July 01, 2022
Correspondence: Eugenio Américo Palomino Portilla.
Address: Doña Victoria Street, C-12, Doña Victoria Urbanization, Santiago de Surco.
Telephone number: 998 483195
E-mail: eugenio.palomino@urp.edu.pe
Article published by the Journal of the faculty of Human Medicine of the Ricardo Palma University. It is an open access article, distributed under the terms of the Creatvie Commons license: Creative Commons Attribution 4.0 International, CC BY 4.0(https://creativecommons.org/licenses/by/1.0/), that allows non-commercial use, distribution and reproduction in any medium, provided that the original work is duly cited. For commercial use, please contact revista.medicina@urp.edu.pe.
REFERENCES
