CASO CLÍNICO
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2023 - Universidad Ricardo Palma10.25176/RFMH.v23i2.5656
ENFERMEDAD ULTRARARA FRECUENTE EN PERÚ:
REPORTE DE CASOS DEL SÍNDROME
FATCO
ULTRARARE DISEASE FREQUENT IN PERU: CASE REPORTS OF FATCO SYNDROME
Wendy Gutierrez-Baca
 1,2,
D'Karlo Dongo-Cornejo
1,
1,2,
D'Karlo Dongo-Cornejo
1,
Hugo H. Abarca Barriga
1,3
1 Carrera Profesional de Medicina Humana, Universidad Científica del Sur. Lima, Perú.
2 CHANGE Research Working Group, Facultad de Ciencias de la Salud, Carrera de Medicina Humana, Universidad Científica del Sur. Lima, Perú.
3 Servicio de Genética & Errores Innatos del Metabolismo. Instituto Nacional de Salud del Niño-Breña. Lima, Perú.
RESUMEN
El síndrome FATCO (fibular aplasia, tibial camptomelia, oligosyndactyly) está caracterizado por la presencia de anomalías en miembros inferiores. Es una enfermedad, de la cual no se ha precisado la etiología genética hasta la actualidad; sin embargo, se ha planteado que el tipo de herencia es dominante autosómica. La frecuencia de presentación a nivel global es muy rara y esta es la razón principal de los pocos pacientes publicados hasta la fecha.
Existe un reporte de la presentación inusual de catorce pacientes peruanos, diagnosticados en un solo centro, con las características clínicas del síndrome FATCO en un período de 13 años. A la fecha, se han publicado catorce pacientes a nivel mundial, con los cuales se comparó y discutió los datos clínicos y radiológicos. Además, se analizaron las características demográficas, antecedentes familiares, sexo, edad y anomalías concomitantes.
Palabras Clave: Aplasia de fíbula, campomelia de tibia, oligodactilia, síndrome FATCO. (Fuente: DeCS – BIREME).
ABSTRACT
The fibular aplasia, tibial campomelia, oligosyndactyly (FATCO) syndrome is characterized by the variable leg anomalies. The genetic etiology of this disease has not been determined to date; however, it has been suggested that the genetic inheritance is autosomal dominant. The frequency of presentation globally is infrequent and this is the main reason for the low number of patient reports.
There’s a report of the unusually high presentation of 14 Peruvian patients diagnosed at a single center with the clinical features of FATCO syndrome over a 13-year period. We compare and discuss the clinical and radiological data of our patients with those of the 14 cases described worldwide. In addition, the demographic characteristics, family history, sex, age, and concomitant anomalies are analyzed.
Keywords: Fibular aplasia, tibial campomelia, oligosyndactyly, FATCO syndrome. (Source: MeSH – NLM).
INTRODUCCIÓN
El síndrome FATCO (del inglés Fibular Aplasia, Tibial Camptomelia, Oligosyndactyly) se caracteriza por la presencia de aplasia o hipoplasia de fíbula, campomelia de tibia y oligosindactilia (1). Las manifestaciones clínicas son muy variables, desde deficiencias en las cuatro extremidades, acompañado de malformaciones cardíacas hasta anomalías en sólo el miembro inferior con las tres características mínimas básicas (2,3,4). Otros síndromes que tienen aplasia fibular son síndrome Du Pan, síndrome Al-Awadi/Raas- Rotschild y síndrome Fémur-Fíbula-Ulna (5). Respecto al signo de campomelia en huesos largos, este se puede presentar también en diferentes entidades tal como el síndrome Roberts, displasias esqueléticas, constricción fetal, pseudoartrosis y pterigium de miembros (6).
El síndrome FATCO (codificado en Mendelian Inheritance in Man con #246570) es una enfermedad genética y congénita, de etiología desconocida, no obstante se ha planteado que tiene una herencia de tipo dominante autosómica, con una frecuencia de presentación ultrarara, es decir que tiene una prevalencia menor de 1 en un millón de personas (7,8). Fue descrito por primera vez por Hecht y Scott en 1981, en dos hermanos con deficiencias transversales de miembros (9). Fue Courtens et al. en el 2005, quienes acuñan el término de FATCO, recopilando los dos pacientes estudiados por Hecht y precisando que existen tres casos más, los cuales fueron descritos previamente como otros síndromes (1). Hasta la actualidad y a nivel mundial, solo se cuenta con información de veintidós pacientes (10-24), de los cuales se han reportado 5 casos en Perú previamente, sin embargo, no se tiene la descripción clínica (25).
Describiremos las características clínicas de catorce pacientes peruanos diagnosticados con síndrome FATCO, los cuales compararemos con los pacientes reportados previamente, y también, poder aportar con más información clínica sobre esta enfermedad, el cual tiene gran repercusión en la calidad de vida de las personas afectadas. Por este motivo, al contar con una mayor y mejor descripción clínica se podría comprender mejor la patología. Re saltar que la mayoría de los casos reportados a nivel mundial, se encontrarían en Sudamérica, siendo casi en su totalidad de Perú.
MATERIAL Y MÉTODOS
Por la ausencia de pruebas de diagnóstico genético, criterios clínicos establecidos y al ser una patología poco frecuente, se decidió incluir a todos los pacientes que presentaban por lo menos dos de las tres características clínicas y radiológicas fenotípicas mínimas, que son la presencia de aplasia/hipoplasia de fíbula, campomelia de tibia y oligosindactilia; permitiendo encontrar un mayor número de niños evaluados. Los pacientes registrados fueron desde el 2008 hasta el 2021 atendidos en el Servicio de Genética & Errores Innatos del Metabolismo del Instituto Nacional de Salud del Niño Breña, el cual es uno de los pocos centros de referencia nacional peruano de pediatría y sobre todo de genética pediátrica. Para la recolección de los datos, se obtuvo la aprobación de dos comités de ética locales (Universidad Científica del Sur e Instituto Nacional de Salud del Niño-Breña), el cual permitió mantener la privacidad y la confidencialidad de las historias clínicas. La información recolectada fueron antecedentes prenatales, natales y familiares, antropometría al nacimiento, procedencia de los padres y abuelos, así como el fenotipo de cada paciente.
RESULTADOS
Se registraron 14 pacientes que reunieron los criterios diagnósticos clínicos y radiológicos del síndrome FATCO. (Figura 1)
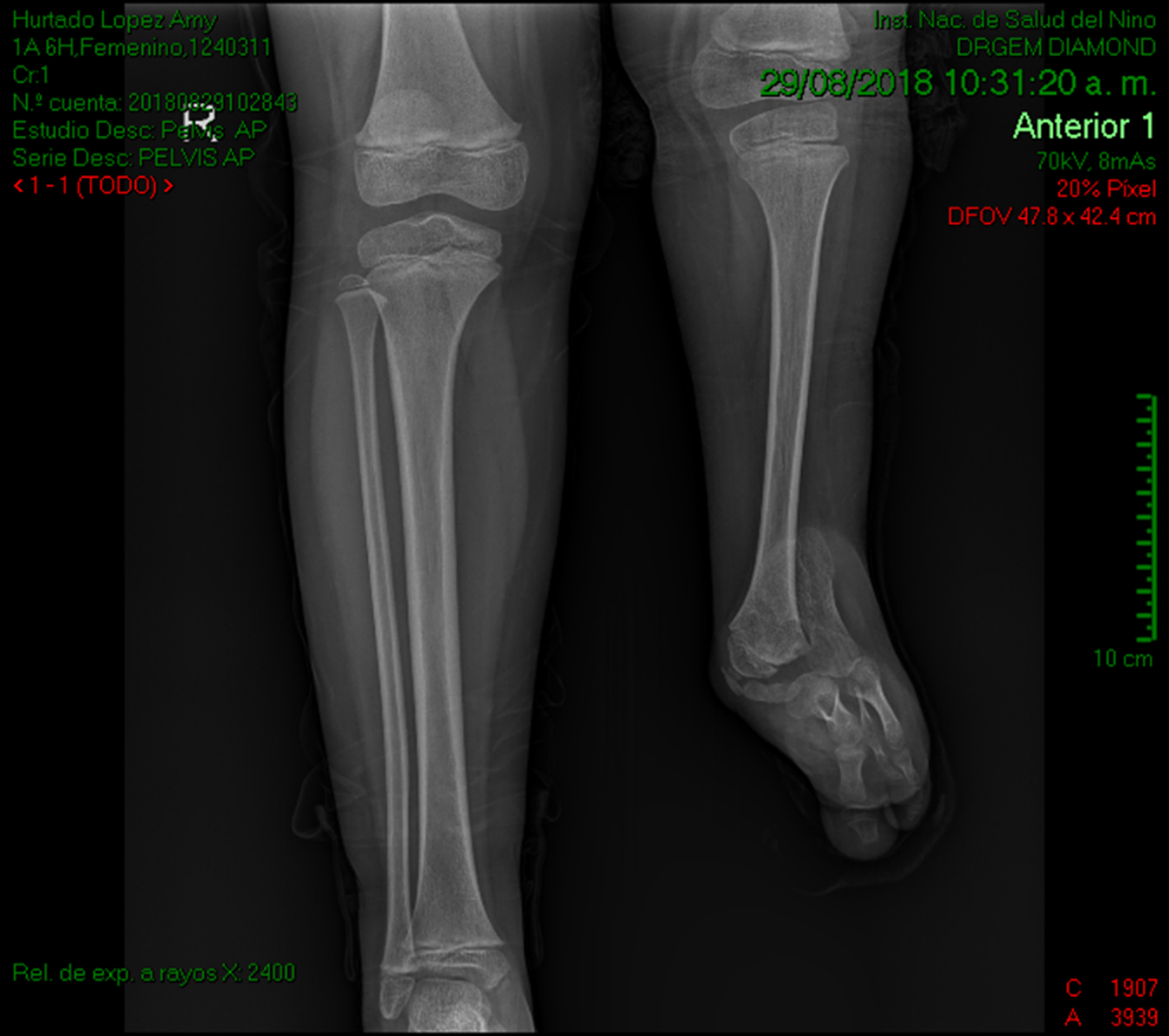
Figura 1. Características clínicas y radiológicas de los pacientes. (A) Radiografía de las extremidades inferiores
que muestra hipoplasia tibial, aplasia fibular y oligosindactilia en la pierna izquierda.
Respecto a la edad, los pacientes tuvieron una mediana de 14 meses (RIC=82,5), las madres con una media de 29 años (σ =8,04) y en los padres con un promedio de 30 años (σ =7,22). El 64% fue de género masculino. Durante el embarazo no se reportó intercurrencias. El lugar de procedencia de los abuelos fue muy variable (Ayacucho, Cusco, Cajamarca, Puno, La Libertad, Lima, Ica, Cerro de Pasco y Huancavelica), no encontrándose alguna región específica con mayor reporte de pacientes. No se observaron antecedentes familiares de anomalías a nivel musculoesquelético, ni de consanguinidad parental. Dentro de los antecedentes natales se obtuvo medidas de tendencia central y de dispersión del peso, talla y perímetro cefálico de 3 262 g (σ = 391 g), 50 cm (σ =1,21 cm) y 34,4 cm (σ =1,77 cm), respectivamente. El Apgar reportado se encontró en rangos normales (Tabla 1).
Tabla 1. Características demográficas de los pacientes del Instituto Nacional de Salud del Niño con síndrome
FATCO en Perú (2008-2021)
|
Características clínicas |
Promedio de resultados |
|---|---|
|
Datos del Paciente |
|
|
Edad del paciente (meses) |
14 (RIC= 82,5) |
|
Sexo |
9/ 14 Masculino |
|
Peso al nacer (g) |
3268,10 (σ=391,88) |
|
Talla al nacer (cm) |
50,69 (σ=1,21) |
|
Perímetro cefálico |
34,43 (σ=1,77) |
|
Apgar |
8/8 Normal |
|
Antecedentes familiares |
|
|
Edad de la madre (años) |
29,3(σ=7,66) |
|
Edad del padre (años) |
30,4(σ=6,89) |
|
Hermanos |
6/ 14 Positivos |
|
Abortos |
4/ 14 Positivos |
|
Antecedentes familiares de malformaciones en miembros |
0 / 14 Positivos |
|
Antecedentes prenatales |
S/P |
|
Consanguinidad |
0/ 14 Positivos |
S/P: sin particularidades, ND: no descrito; NR: No realizado; NE: no especificado; Positivo: presencia de anomalía;
Negativo: No hay presencia de anomalía; RIC: Rango intercuartílico; DE: Desviación
La presencia de campomelia e hipoplasia tibial se halló con similar frecuencia en ambos miembros inferiores (ver Figura 1 y Figura 2, Tabla 2).
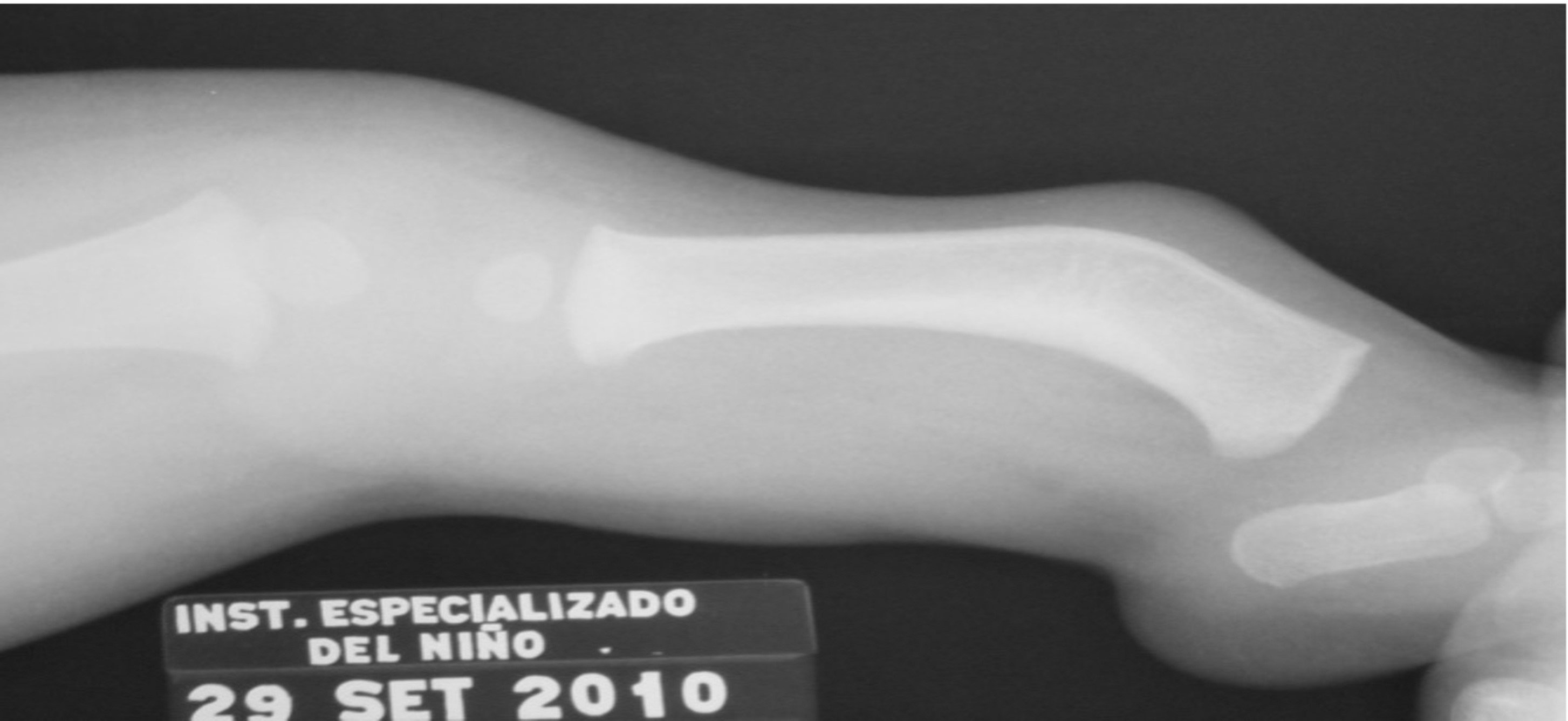
Figura 2. Radiografía de pierna izquierda con campomelia tibial y aplasia de peroné
|
Características clínicas |
POSITIVOS |
NEGATIVOS |
NO ESPECIFICADOS |
% |
|---|---|---|---|---|
|
Anomalías en extremidades superiores |
|
|
|
|
|
Sindactilia completa de mano izquierda 1er- 2do y 4to -5to |
1 |
12 |
1 |
7,14 |
|
Sindactilia cutánea de 3er-5to |
0 |
13 |
1 |
0 |
|
Ausencia de cuarto metacarpiano de mano |
1 |
12 |
1 |
7,14 |
|
Anomalía en manos |
3 |
10 |
1 |
21,42 |
|
Anomalías en extremidades inferiores |
|
|
|
|
|
Hoyuelo en pierna |
8 |
2 |
4 |
57,14 |
|
Ausencia del 5to ortejo |
12 |
1 |
1 |
85,71 |
|
Ausencia de 4to–5to ortejo |
7 |
6 |
1 |
50 |
|
Ausencia de 3er-5to ortejo |
3 |
10 |
1 |
21,42 |
|
Sindactilia de 1er-2do ortejo |
3 |
10 |
1 |
21,42 |
|
Ausencia de fíbula |
13 |
0 |
1 |
92,85 |
|
Campomelia de tibia |
14 |
0 |
0 |
100 |
|
Hipoplasia de tibia |
14 |
0 |
1 |
100 |
|
Otras anomalías |
|
|
|
|
|
Uñas |
0 |
11 |
3 |
0 |
|
Desarrollo psicomotor |
0 |
10 |
4 |
0 |
|
Cribado de anomalías óseas en otras regiones |
0 |
8 |
6 |
0 |
|
Cardiopatía congénita |
0 |
11 |
3 |
0 |
|
RMN cerebral |
0 |
0 |
14 |
0 |
|
Fondo de ojo |
0 |
0 |
14 |
0 |
|
Ultrasonografía renal |
0 |
5 |
9 |
0 |
|
Potenciales evocados auditivos |
0 |
0 |
14 |
0 |
|
Cariotipo |
0 |
0 |
14 |
0 |
S/P: sin particularidades, ND: no descrito; NR: No realizado; NE: no especificado; Positivo: presencia de anomalía;
Negativo: No hay presencia de anomalía
Se observó aplasia fibular de miembro izquierdo en seis pacientes, y del lado derecho en cinco pacientes, e hipoplasia fibular en dos pacientes y no especificado en uno. En un paciente se describió la presencia de oligodactilia, sin precisar el número ni el lado; La oligodactilia que incluyó metatarsos, se observó desde el 3º al 5º ortejo en tres pacientes (21,4%); solo del 5º ortejo en dos (14,3%) y del 4º-5º ortejo oligodactilia en siete pacientes (50%) (Figura 3 - a y b).
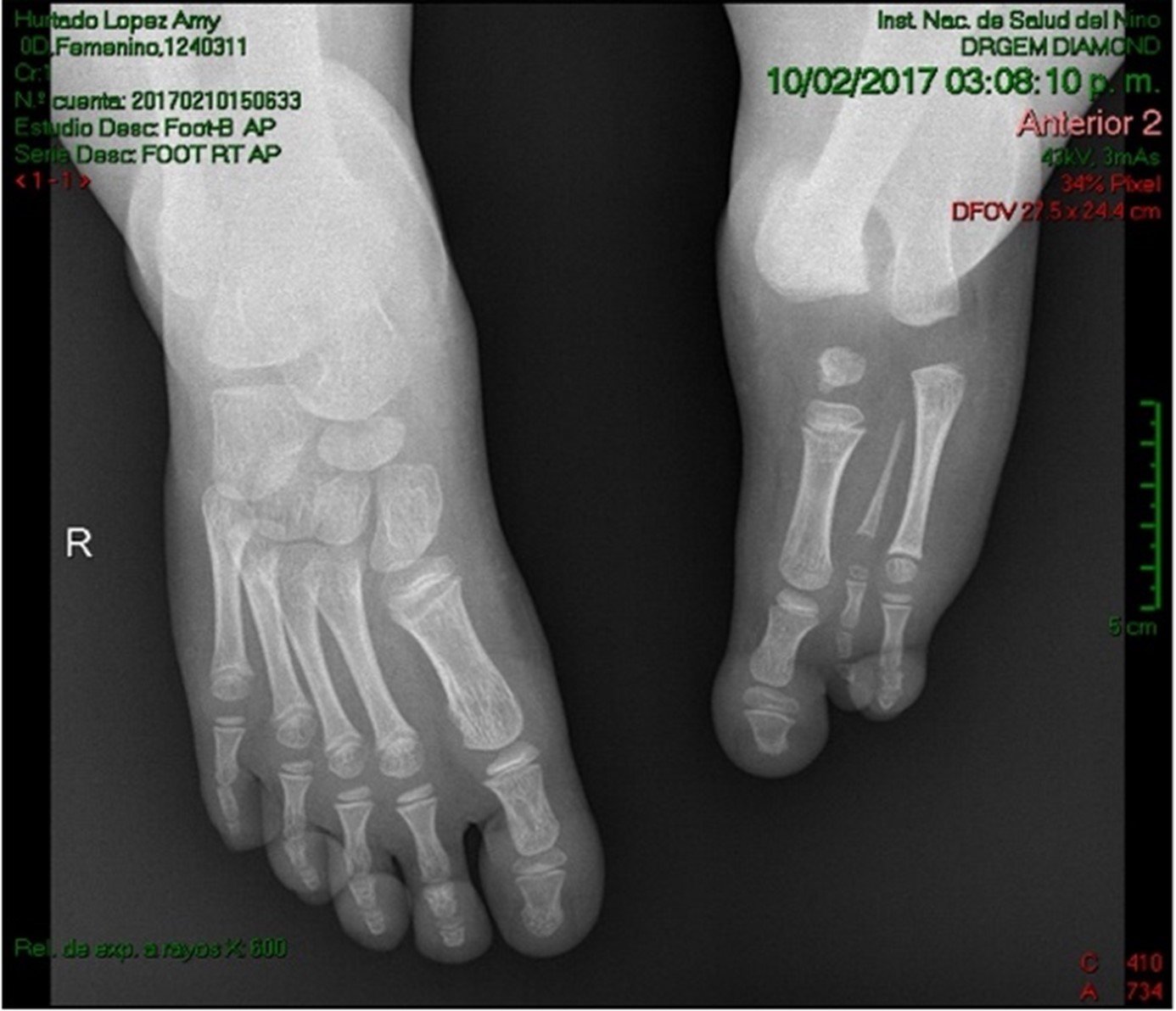 separacion
separacion
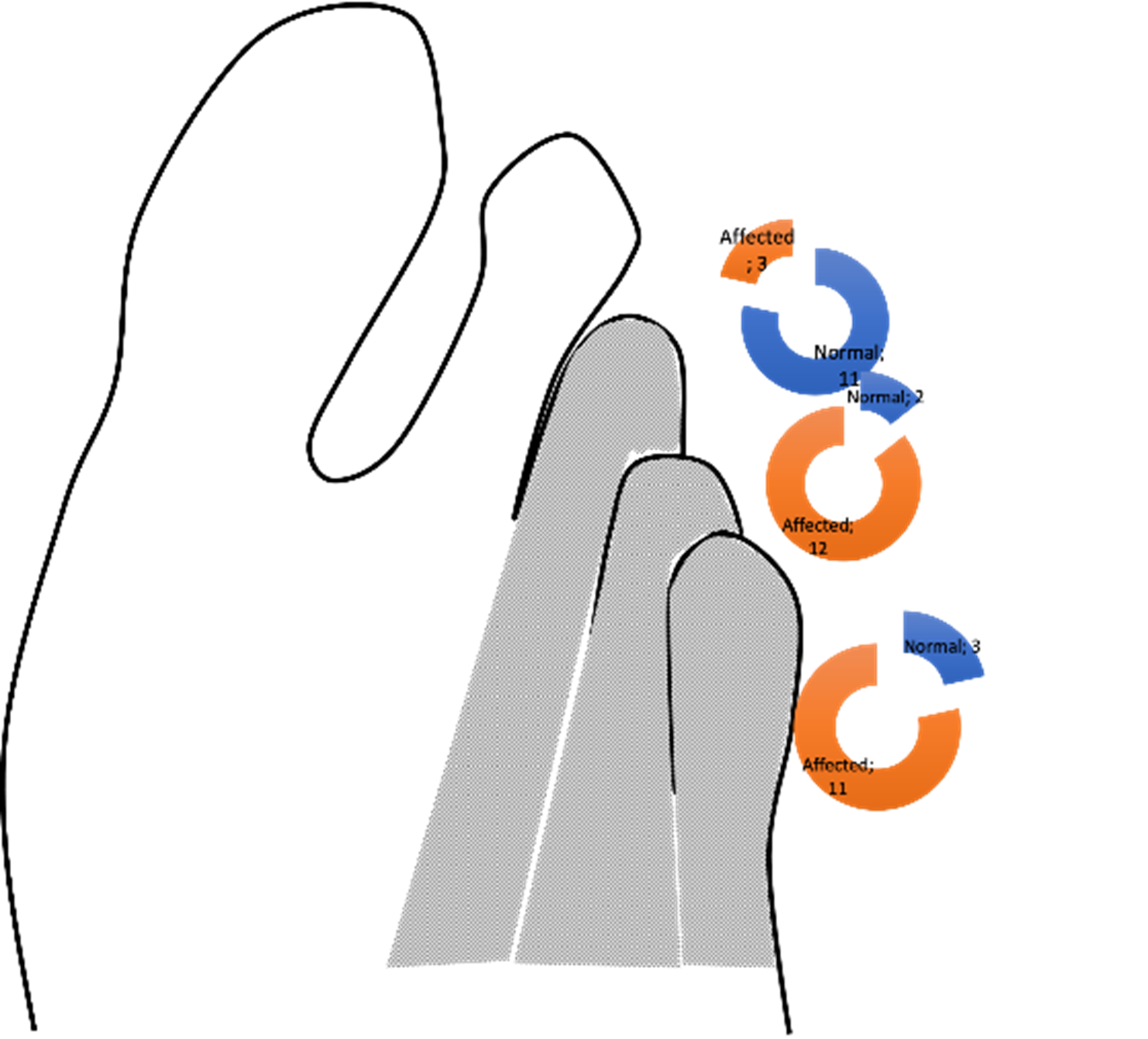
Figura 3. a) Radiografía de ambos pies que muestra oligosindactilia en el pie izquierdo. b) Frecuencia de dedos de los pies afectados en los pacientes descritos.
Se evidenció sindactilia del primer y segundo ortejo en tres pacientes (21,4%). La presencia de hoyuelo en la pierna se presentó en el lado derecho en cinco pacientes (35,7%) y el izquierdo en tres pacientes (21,4%) (Figura 4). El desarrollo psicomotor fue normal en todos los pacientes.
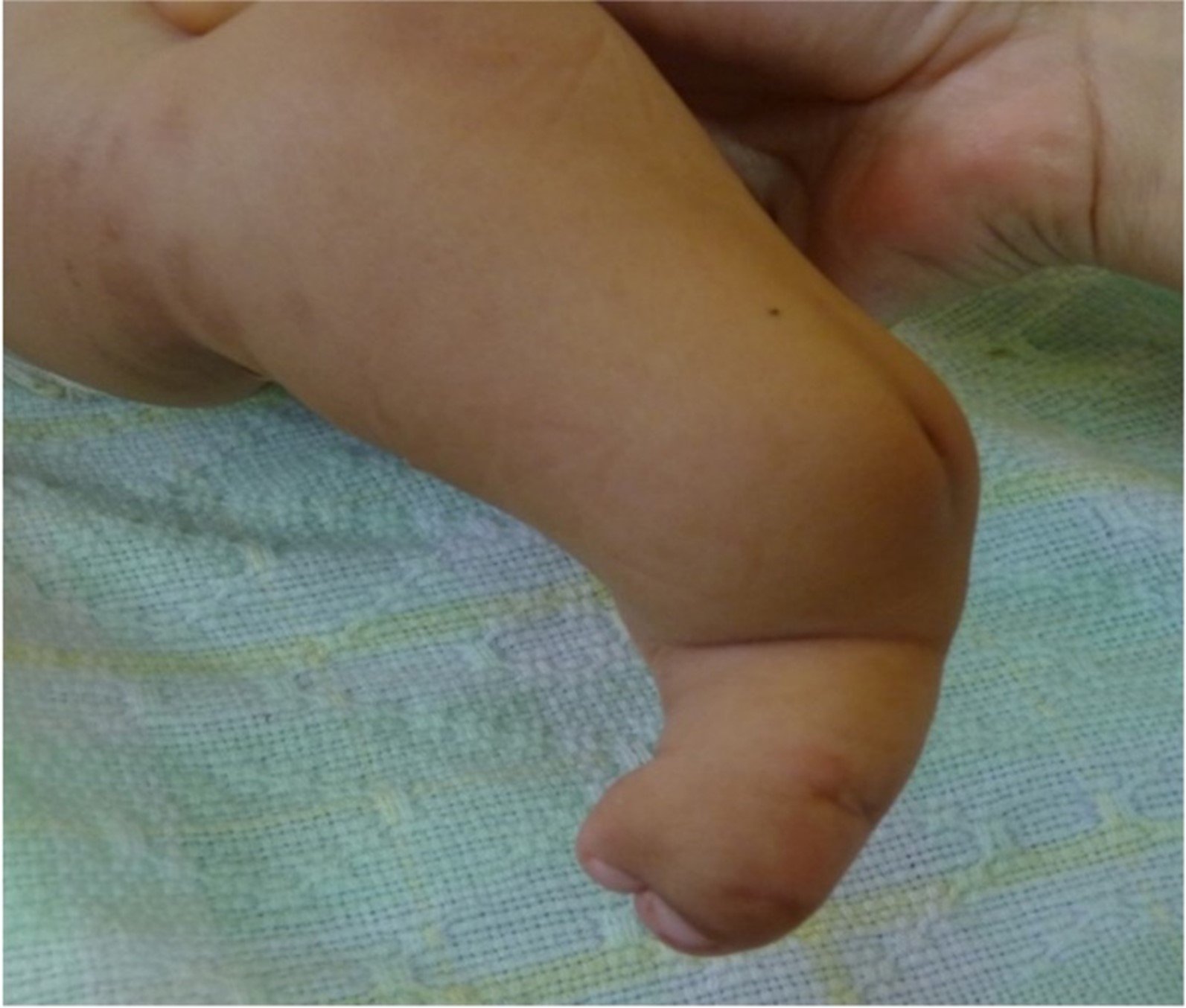
Figura 4. Fotografía de pierna pierna izquierda con hoyuelo (flecha blanca) y oligosindactilia.
En la mano, se evidenció anomalías en tres pacientes (21,4%), como la presencia de agenesia de pulgar en dos pacientes (14,2%) y del quinto dedo en uno (7,1%), sindactilia de mano izquierda entre el 1º y 2º dedos, así como entre el 4º-5º dedos en un paciente (7,1%) y ausencia del cuarto metacarpiano en uno (7,1%). (Figura 5 y Tabla 2).
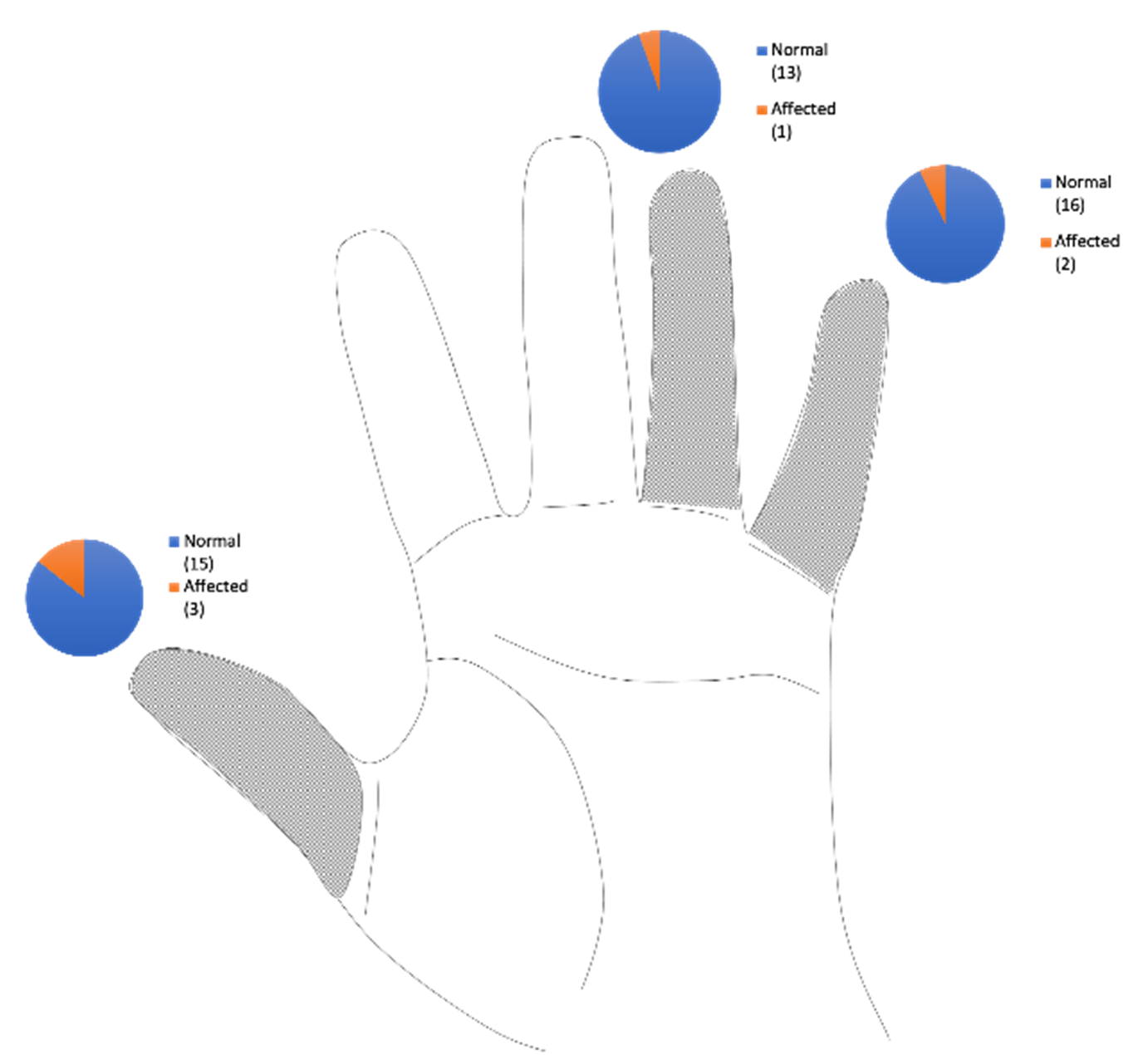
Figura 5. Frecuencia de dedos afectados en los pacientes descritos.
DISCUSIÓN
Los catorce pacientes peruanos representan el 50% de todos los pacientes reportados en el mundo hasta la fecha (10-24). Las anomalías en las extremidades superiores se observaron en el 21,4% de los casos nacionales, mientras que en los casos previos se evidenció en 2 de cada 3 pacientes reportados hasta el momento (64,2%)
Los primeros informes sobre esta investigación afirman que la frecuencia del número total de anomalías era la misma en ambas piernas. La presencia de hoyuelos en las piernas en esta
revisión prevaleció en el miembro inferior derecho (35,7% vs. 21,4%) y en reportes de casos previos se observa similar frecuencia.
Con relación a la oligodactilia, a nivel nacional, se observó con más frecuencia el compromiso del 4º-5º ortejo, mientras que a nivel internacional la frecuencia fue mayor en el
quinto ortejo (Tabla 3) (7-22). La sindactilia del primer y segundo ortejo fue poco descrita en los pacientes, pero
el lado afectado fue únicamente el derecho en este reporte y anteriores manuscritos.
Tabla 1. Características demográficas de los pacientes del Instituto Nacional de Salud del Niño con síndrome
FATCO en Perú (2008-2021)
|
Características clínicas |
POSITIVOS |
NEGATIVOS |
NO ESPECIFICADOS |
% |
|---|---|---|---|---|
|
Anomalías en extremidades superiores |
|
|
|
|
|
Sindactilia completa de mano izquierda 1er-2do y 4to-5to |
3 |
9 |
3 |
21,4 |
|
Sindactilia cutánea de 3er-5to |
2 |
10 |
2 |
14,3 |
|
Ausencia de cuarto metacarpiano de mano |
4 |
8 |
2 |
28,6 |
|
Alteración en Manos |
5 |
6 |
3 |
35,7 |
|
Anomalías en extremidades inferiores |
|
|
|
|
|
Hoyuelo en pierna |
11 |
1 |
2 |
78,6 |
|
Ausencia de 5to ortejo |
13 |
1 |
0 |
92,9 |
|
Ausencia de 4to-5to ortejo |
6 |
8 |
0 |
42,9 |
|
Ausencia de 3er-5to ortejo |
1 |
13 |
0 |
7,1 |
|
Sindactilia de 1er-2do ortejo |
3 |
11 |
0 |
21,4 |
|
Ausencia de fíbula |
14 |
0 |
0 |
100 |
|
Campomelia de tibia |
14 |
0 |
0 |
100 |
|
Hipoplasia de tibia |
12 |
2 |
0 |
85,7 |
|
Otras anomalías |
|
|
|
|
|
Uñas |
1 |
7 |
6 |
7,1 |
|
Desarrollo psicomotor |
0 |
9 |
5 |
0 |
|
Cribado de anomalías óseas en otras regiones |
3 |
5 |
6 |
21,4 |
|
Cardiopatía congénita |
1 |
9 |
4 |
7,1 |
|
RMN cerebral |
1 |
5 |
8 |
7,1 |
|
Fondo de ojo |
0 |
4 |
10 |
0 |
|
Ultrasonografía renal |
0 |
7 |
7 |
0 |
|
Potenciales evocados auditivos |
0 |
4 |
10 |
0 |
|
Cariotipo |
1 |
3 |
10 |
7,1 |
S/P: sin particularidades, ND: no descrito; NR: No realizado; NE: no especificado; Positivo: presencia de anomalía;
Negativo: No hay presencia de anomalía
La ausencia del quinto metatarsiano y sus respectivas falanges prevaleció en el pie derecho en los pacientes peruanos, comparado a previos informes, en los cuales predominó en el lado izquierdo. Respecto a la ausencia del cuarto metatarsiano, esta característica predomina en este reporte, presentando en el 85,7%. Sin embargo, en previas descripciones, esta característica aparece en 42,8% de los pacientes y la ausencia del tercer metatarsiano no fue un hallazgo significativo en los casos reportados.
Previos reportes, así como en Perú, se describió la presencia de aplasia fibular predominando en la pierna derecha, que se asoció en todos los pacientes a campomelia tibial. Investigaciones previas, muestran que la hipoplasia tibial se describió en el 85,7% de los casos reportados, con afectación bilateral en el 28,6%, asimismo en este estudio se observó en todos los pacientes sin predominio de un miembro inferior.
Previamente se mostró un paciente con uñas hipoplásicas, no observándose esta anomalía en ningún paciente peruano. De la misma forma el cribado de anomalías óseas en otras regiones, solo se presentaron en los pacientes internacionales, siendo no prevalentes en nuestro medio. Y con relación al desarrollo psicomotor, solo se halló afectado el retraso en la marcha, mientras que en los pacientes peruanos no se observó (26).
El síndrome de FATCO es una enfermedad genética ultra rara, donde una de sus limitaciones es la falta de información acerca de esta patología, como la etiología; Un estudio publicado recientemente comenta sobre la posible etiología de este síndrome, comentándola como un desafío ya que no se trataría de solo una alteración monogénica sino posiblemente algo más complejo como un trastorno del desarrollo vascular, variantes somáticas o un mosaicismo de bajo nivel en la sangre, para lo cual sería necesario un avance en las herramientas tecnológicas para su diagnóstico, siendo por ahora inexacta (27). La otra limitación encontrada es en base a la ausencia de estudios moleculares para el diagnóstico de síndrome de FATCO, y además existen enfermedades genéticas que tienen algunas de las características clínicas, pero que se pudo diferenciar mediante el uso de herramientas diagnósticas, como el uso de portales como el OMIM (On line Mendelian Inheritance in Man-www.omim.org-) o programas de búsqueda como el Face2Gene, Phenomizer o Possum (28-29).
Sin embargo, esta limitación no afecta al estudio, ya que nos llevó a incorporar el detalle fenotípico de cada paciente. Y, por último, el síndrome FATCO no tiene una prueba gold-standar para el diagnóstico, el cual, si hubiera, facilitaría el diagnostico en cualquier estadio (prenatal y postnatal); sin embargo, es necesario considerar los diagnósticos diferenciales que muchos de ellos tienen una etiología genética identificada a través de las pruebas de secuenciación masiva (30-31).
No hemos podido establecer según lo reportado hasta el momento, la razón del porque en Perú existiría un mayor número de pacientes; sin embargo, existe una leyenda en la selva peruana sobre la existencia de un ser a quién denominan como Chullachaqui, quien tiene miembros inferiores desiguales, el derecho grande y el izquierdo chico; por lo que deduciríamos que esta condición ha sido vista desde hace mucho tiempo en esta población (32). En este sentido, es necesario realizar estudios genómicos como la secuenciación masiva o la utilización de micromatrices con un mayor número de marcadores con la finalidad de detectar variantes en un único nucleótido, de múltiples nucleótidos o variantes en el número de copias lo cual servirá para establecer no sólo el diagnóstico molecular etiológico; sino, también, de reconocer los procesos de la diferenciación embrionaria de los miembros, establecer el riesgo de recurrencia familiar e individual y plantear mejoras terapéuticas futuras. Por otro lado, si estos estudios no fuesen concluyentes se tendría que realizar estudios epigenéticos, así como estudio de expresión (transcriptoma) entre otros (31-32).
Contribuciones de Autoría:
Los autores participaron en la génesis de la idea, diseño del proyecto, desarrollo, recolección e interpretación de data, análisis de resultados y preparación del manuscrito.
Financiamiento:
Autofinanciado.
Declaración de conflictos de intereses:
Los autores declaran no tener conflicto de interés en la publicación de este artículo.
Recibido:
10 de febrero del 2023.
Aprobado:
27 de abril del 2023.
Correspondencia:
Hugo Abarca Barriga.
Dirección:
CP Medicina Humana. Universidad Científica del Sur.
Teléfono:
979 301 132
Correo electrónico:
habarca@cientifica.edu.pe
Artículo publicado por la Revista de la Facultad de Medicina Humana de la Universidad Ricardo Palma. Es un articulo de acceso abierto, distribuido bajo los términos de la Licencia Creatvie Commons: Creative Commons Attribution 4.0 International, CC BY 4.0(https://creativecommons.org/licenses/by/1.0/), que permite el uso no comercial, distribucion y reproducción en cualquier medio, siempre que la obra original sea debidamente citada. Para uso comercial, por favor póngase en contacto con revista.medicina@urp.edu.pe.
REFERENCIAS BIBLIOGRÁFICAS
