CLINICAL CASE
REVISTA DE LA FACULTAD DE MEDICINA HUMANA 2023 - Universidad Ricardo Palma10.25176/RFMH.v23i4.5814
FIRST CASE OF THANATOPHORIC DYSPLASIA TYPE 1 IN THE PERUVIAN ANDES WITH PATHOGENIC VARIANT IN THE FGFR3 GENE.
PRIMER CASO DE DISPLASIA TANATOFÓRICA TIPO 1 EN LOS ANDES PERUANOS CON VARIANTE PATOGÉNICA EN EL GEN FGFR3
Torres-Salinas Carlos
 1,2,a
1,2,a
Ledesma-Porras Yesenia
1,3,b
1 Faculty of Human Medicine. Continental University, Peru.
2 Bicentennial EsSalud Jauja Hospital, Peru.
3 National Ramiro Prialé EsSalud Hospital, Peru.
a Pediatrician.
b Geneticist.
ABSTRACT
Thanatophoric dysplasia type 1 is a form of lethal skeletal dysplasia, characterized by disproportions of the axial-appendicular skeleton in addition to short stature, macrocephaly, frontal prominence, narrow chest, femoral bowing, and micromelia. These phenotypic characteristics are the result of pathogenic variants in the fibroblast growth factor receptor 3 gene (FGFR-3), located on chromosome 4p16.3. For its study, obstetric ultrasound, physical examination, and radiographic findings are important. However, the diagnosis must be confirmed by genetic study in order to discover new variants or associations, as well as to make known its real casuistry in a given region.
Keywords: Skeletal dysplasia, thanatophoric dysplasia, fibroblast growth factor receptor type 3. (Source: DeCS – BIREME)RESUMEN
La displasia tanatofórica tipo 1 es una forma de displasia esquelética letal, se caracteriza por desproporciones del esqueleto axial-apendicular además de talla baja, macrocefalia, prominencia frontal, tórax estrecho, arqueamiento femoral y micromelia. Dichas características fenotípicas son el resultado de variantes patogénicas en el gen del receptor 3 del factor de crecimiento de fibroblastos (FGFR-3), localizada en el cromosoma 4p16.3
Para su estudio la ecografía obstétrica, el examen físico y los hallazgos radiográficos son importantes. Sin embargo, se debe confirmar el diagnóstico mediante estudio genético a fin de descubrir variantes o asociaciones nuevas, así como dar a conocer su casuística real en una determinada región.
Palabras clave: Displasia esquelética, displasia tanatofórica, receptor tipo 3 de factor de crecimiento de fibroblastos. ( Fuente: DeCS – BIREME)Thanatophoric dysplasia (TD) is a severe skeletal dysplasia caused by pathogenic variants in the FGFR3 gene (fibroblast growth factor receptor 3). The most common pathogenic variants occur in the extracellular domain, either by a cysteine to arginine substitution at position 248 (Arg248Cys) or a cysteine to tyrosine substitution at position 373 (Tyr373Cys), while for thanatophoric dysplasia type 2 it is (Lys650Glu). TD type I presents with micromelia with femoral bowing and craniosynostosis of varying degrees, while TD type II is characterized by micromelia with straight femurs, the presence of moderate to severe craniosynostosis, and a "cloverleaf" skull deformity (1).
Regarding skeletal dysplasias in Latin America, the largest study shows a prevalence of 3.2 cases per 10,000 live births (LB) (2). Meanwhile, thanatophoric dysplasia is even rarer with 0.37 cases per 10,000 LB (3).
To date, there are no published cases of thanatophoric dysplasia type 1 in Peru with confirmation by early skeletal dysplasia panel study and subsequently reported to the scientific community.
We present a case of thanatophoric dysplasia type I diagnosed by detecting the FGFR3 gene mutation in the central region of Peru, and informed consent has been obtained for the exclusive medical purposes of publication.
A 38-week-old male neonate was born via elective cesarean section due to skeletal malformations and polyhydramnios diagnosed by ultrasound at 36 weeks (previous private ultrasound studies showed no apparent abnormalities). The maternal age was 40 years and the paternal age 46 years, G4P3003, VDRL: non-reactive, HIV: non-reactive, and complete prenatal controls in a rural health center.
Further history taking revealed no evidence of inbreeding, drug, alcohol, or toxic substance use. Also, the parents did not exhibit any current illnesses, and there were no family histories of congenital diseases.
At birth, the patient required tactile stimulation, aspiration of secretions, and one cycle of positive pressure ventilation with a T-piece (Neopuff®), APGAR score: 61-85, umbilical cord arterial blood gas: pH: 7.18, PaO2: 55.1 mmHg, PaCO2: 52 mmHg, HCO3: 15.8 mmol/L, EB: -9.8 mmol/L, lactate: 4.5 mmol/L, SatO2: 85%, Silverman-Anderson scale: 5 points. This led to the decision to switch to CPAP (nasal) support, which partially improved respiratory difficulty and SatO2: 92%. At 8 hours, the device was switched to a high-flow cannula with an acceptable response.
In the anthropometric evaluation, the following measurements were found: weight: 2710g, length: 39 cm, head circumference: 36 cm, thoracic circumference: 29.5 cm. Additionally, craniofacial dysmorphism, relative macrocephaly, short neck, narrow chest, short stature, accordion-like skin, rhizomelic shortening of all four limbs, brachydactyly, and generalized hypotonia were observed (Fig. 1).

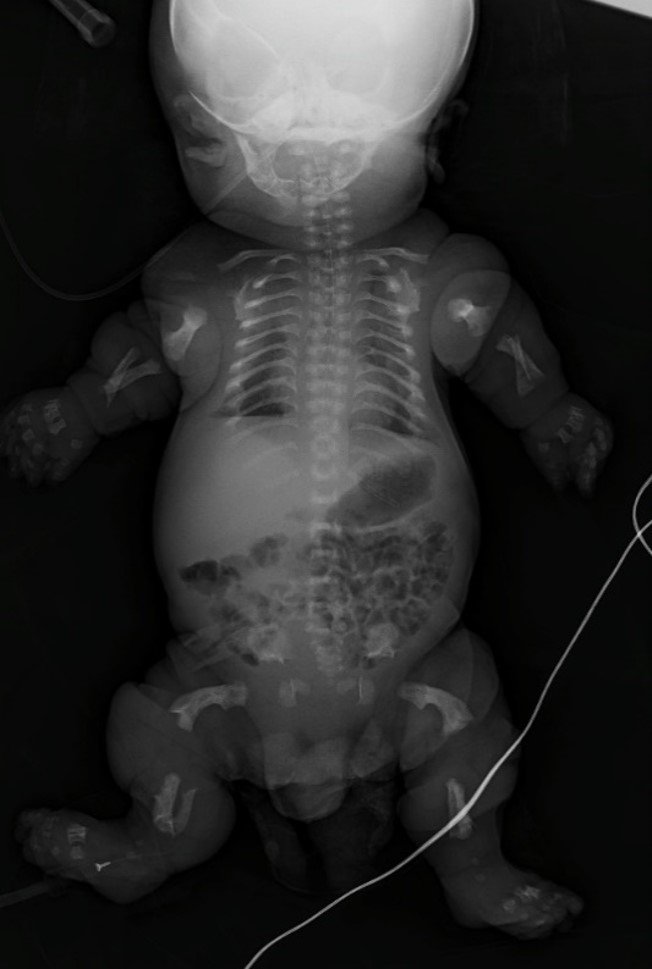
The radiographic study showed a small cranial base, platyspondyly, rib arcs in a "bell tower" arrangement related to a narrow chest, hypoplastic ilium, and femoral bowing, with the classic "telephone receiver" finding (Fig. 2).
Additionally, a genetic panel for skeletal dysplasias including 358 genes was performed, identifying 01 pathogenic variant: in the FGFR3 gene, variant c.1108G>T (p.Gly370Cys), (NM_000142.4). Incidentally, a variant c.2656C>T (p.Arg886*) was found in the KIAA0753 gene (NM_014804.2), which is associated with autosomal recessive skeletal ciliopathies. This variant has not been reported in the literature in individuals affected with conditions related to KIAA0753 (Fig. 3).
The clinical evolution showed progressive deterioration over time, and despite various oxygenation and ventilation support devices, the patient died at 16 days due to respiratory arrest secondary to pulmonary hypoplasia and significant narrowing of the thoracic cage.
DISCUSSION
Thanatophoric dysplasia (TD) is an autosomal dominant disorder, typically caused by a de novo pathogenic variant. It is part of a subgroup related to skeletal chondrodysplasias, which are divided into 02 types (although they do not always correlate with the found gene); these are TD type I with curved "telephone receiver" femurs, platyspondyly, usually a normal skull, and TD type II with straight femurs and a "cloverleaf" skull; phenotypically they show shortening of all extremities, redundant skin folds, depressed nasal bridge, large abdomen, and narrow chest associated with poor pulmonary development. In relation to TD1, like TD2, it is caused by a heterozygous mutation in the gene encoding fibroblast growth factor receptor 3 (FGFR3; 134934) on chromosome 4p16, with OMIM code #187600 (1, 4).
Regarding diagnosis, it is recommended that it be early and antenatal given its prognosis and outcome; the method used is obstetric ultrasound, and the criteria to consider are: severe rhizomelic micromelia with bowing of the lower extremities below the 3rd percentile (p3) for gestational age; hypoplastic thorax determined by a cardiac circumference greater than 60% of the thoracic circumference. (6)
Other findings related to stages of gestation describe that in the first trimester, between weeks 12 and 14, an increase in nuchal translucency and shortening of long bones can be seen, while in the second trimester it is possible to identify the growth deficiency of limbs below the 5th percentile (p5). Platyspondyly, ventriculomegaly, a narrow thoracic cavity with short ribs, polyhydramnios, and femoral bowing can also be observed, especially in TD type I. Meanwhile, the cloverleaf skull and craniosynostosis affecting the coronal, lambdoid, and sagittal sutures giving the skull a trilobed appearance, also known as “Kleeblattschädel,” are more present in TD type II. (1, 5, 7)
However, the evaluation of the aforementioned findings is not always straightforward, which is why, in case of doubt, it is recommended to supplement the ultrasound study using indices such as the ratio: (biparietal diameter / femur length). This is a useful and objective tool for more accurately detecting thanatophoric dysplasias, especially in the early stages of gestation. (8, 9)
Other ultrasound findings, such as temporal lobe dysplasia, could strengthen the diagnostic suspicion of facing a case of TD; while postnatal morphometric findings could be applied to the antenatal ultrasound search for characteristics of TD, thus optimizing the specificity of obstetric ultrasound. (10)
TDs are a disorder of autosomal dominant inheritance, sporadic with 100% penetrance and low recurrence; most cases are only reported in the affected individuals, with the parents being healthy. Exceptionally, a case of mosaicism has been described in an adult person with asymmetric limbs, congenital hip dysplasia, focal areas of bone bowing, "S"-shaped humerus, extensive acanthosis nigricans, and demise at 30 weeks of a pregnancy with severe skeletal dysplasia and pulmonary hypoplasia. (11)
We must not forget that the FGFR3 gene is responsible for a variety of chondrodysplasias, so cases of hypochondroplasia and pseudoachondroplasia can also be found, ranging from mild to severe, such as Crouzon or Pfeiffer syndrome. (12)
In our case, the pregnant woman followed her obstetric controls in a rural health center without apparent alterations (where there is no full access to trained personnel or ultrasound equipment). The ultrasound controls, as she reports, were carried out privately and without findings of anomalies, of which we had no physical evidence.
At 36 weeks, she was referred to the hospital to complete gestation monitoring according to national regulations, and it was there that ultrasound findings suggestive of TD were detected; upon birth, the baby required initial medical assistance, being dependent on oxygen support and experiencing poor evolution, leading to death at 16 days of life.
The definitive diagnosis was made through genetic study, and incidentally, the coexistence of the heterozygous variant of the KIAA0753 gene was detected. We consider this to have no clinical relevance due to the presence of only a single mutation in this gene, hence not resulting in the characteristic expression of skeletal ciliopathies. Otherwise, it would have been difficult for the clinician to distinguish between the two entities.
In recent years, Peru has been one of the fastest-growing economies in the region. However, we observe a dissociation between this growth and the ability of our hospitals to conduct genetic tests, as in the case described. This constitutes a barrier to access not only to genetic studies but also to rare diseases, and therefore we recommend that this issue should not be taken lightly.
Authorship contributions:
The authors have managed the article in its entirety
Financing:
None.
Declaration of conflict of interest:
None.
Recevied:
July 18, 2023
Approved:
November 2, 2023
Correspondence author:
Dr. Carlos Torres Salinas.
Address:
-
Phone:
+51 947459408
E-mail:
ctorress@continental.edu.pe
Article published by the Journal of the faculty of Human Medicine of the Ricardo Palma University. It is an open access article, distributed under the terms of the Creatvie Commons license: Creative Commons Attribution 4.0 International, CC BY 4.0 (https://creativecommons.org/licenses/by/1.0/), that allows non-commercial use, distribution and reproduction in any medium, provided that the original work is duly cited. For commercial use, please contact revista.medicina@urp.edu.pe.
BIBLIOGRAPHIC REFERENCES
